Fundamentals of Laboratory Management

Overview
Fundamentals of Laboratory Management is designed to serve as an introductory guide to laboratory, quality and regulatory system in laboratories for biotechnicians.
Introduction to Lab & Quality Management System
Laboratory or lab is a place where experiments are carried for the purpose of enhancing scientific knowledge or translating the acquired knowledge for the welfare of mankind[1].
This chapter is designed to serve as an introductory guide to laboratory and quality system in laboratories for biotechnicians. Biotechnicians are personnel that work in laboratories and help scientists/ researchers on various projects. Their role in the lab may include set-up, maintenance, preparing reagents, operating equipment, assisting in experiments, data recording and analysis. It is important for biotechnicians to understand how a laboratory functions, what are the different types of labs and how quality system plays an important role in maintaining consistency in laboratory as well as a framework for quality. Quality has to be built into the processes and protocols in the labs to generate accurate and precise results. Labs can be divided into three categories based on the type of the research that is being conducted: Basic research, translational research, and clinical research.
Basic (pure or fundamental) research is driven by a scientist’s curiosity to find answer to a scientific question. Basic research aims to formulate theory that explain research findings and enhance fundamental understanding of a scientific area. For example, a lab working on basic science question may be determining genetic code of a newly found species of bacteria or studying specific proteins related to the life cycle of SARS-CoV-2 virus that causes coronavirus diseases (COVID).
Translational research implements a ‘bench-to-bedside’ approach. It aims to translate the basic scientific discoveries and research results into medical diagnosis and treatment practices for patients. Translational research involves the use of the fundamental knowledge (a majority of which is gained through the basic sciences) towards practical applications such as developing diagnosis methods or candidate molecules that could serve as therapeutic drug for diseases. For example, the development COVID vaccine is based on the in depth understanding of biology of the virus.
Clinical research involves development and evaluation of the efficacy and safety of new treatments, medications, and diagnostic techniques in patients. The research may enroll volunteers or patients to evaluate or test new medications or interventions. For example, the vaccine produced by Pfizer to prevent COVID was tested in more than 40,000 volunteers before being deemed safe for use on the general public. Clinical research may focus on a medication, medical devices or new therapeutic approaches (treatment research), preventing a disorder or disease from developing or returning (Prevention Research), developing new or better identification techniques (Diagnostic Research), detect diseases or disorders (Screening Research), or prediction of disorders by identifying the genes and biomarkers responsible for certain diseases (Genetic Studies). (www.fda.gov)
1. Types of Lab
Research labs can be found in universities or private companies. In general, there are 4 different types of laboratories.
- Research labs in university generally focus on addressing a question or a hypothesis related to basic science or translational research.
- Research labs in companies address aspects related to commercializing scientific discoveries and processes for making products such as drugs or pharmaceutical and improving services such as diagnostics.
- Core labs support the research of others by providing specific services. For example, using a specific type of assay or in an area of expertise. An example of a core lab is a transgenic mouse core lab provides support services to generate and facilitate the use of mouse models for various diseases.
- Clinical labs are service-based entities involved in analyzing clinical samples. The majority of work in these labs supports patient care and clinical trials. For example, a clinical lab may study patient samples to study the effectiveness of the antibiotics that are prescribed to them.
These labs are described in details in the next section.
Research Labs in Universities: In a large university, there can be different schools focusing on areas such as medicine, nursing, dentistry , law, etc. For example, University of Maryland at Baltimore is a well-recognized public research institute in the nation. It has 7 schools, of which one is the School of Medicine. The School of Medicine has 25 departments, such as biochemistry and molecular biology, family medicine, neurosurgery etc. It also has 10 Research Centers such as Center for Integrative Medicine or Center for Vaccine Development and Global Health. It houses 2 Institutes that work on genome sciences and human virology. These department, centers and institutes may have more than 100 labs working on different scientific areas related to basic, translational, or clinical research. Biotechnicians or laboratory technicians are needed in each and every lab to assist in multiple projects.
The labs in university or academia are usually structured. A lab is usually headed by an individual called the “Principal Investigator” (PI). A PI may have a PhD and/or MD, and not only decides the type of research conducted, but even specific research problems. For example, a PI who has a degree in biology may be working on bacterial genomics and evolution or development of new vaccination strategies. The PI has to apply for grants from time to time to obtain funding money for supporting their research as well as the research staff in their lab from appropriate government agencies (such as National Institute of Health[1]), foundations (such as Bill and Melinda Gates Foundation[2], Burroughs Welcome Fund[3]) or private companies. A big lab may have the primary research staff that includes assistant professors, visiting faculty, and postdoctoral fellows. These lab personnel typically have PhDs or MDs and are semi-independent. They are capable of handling independent projects within the lab, although they may be reporting to the PI.
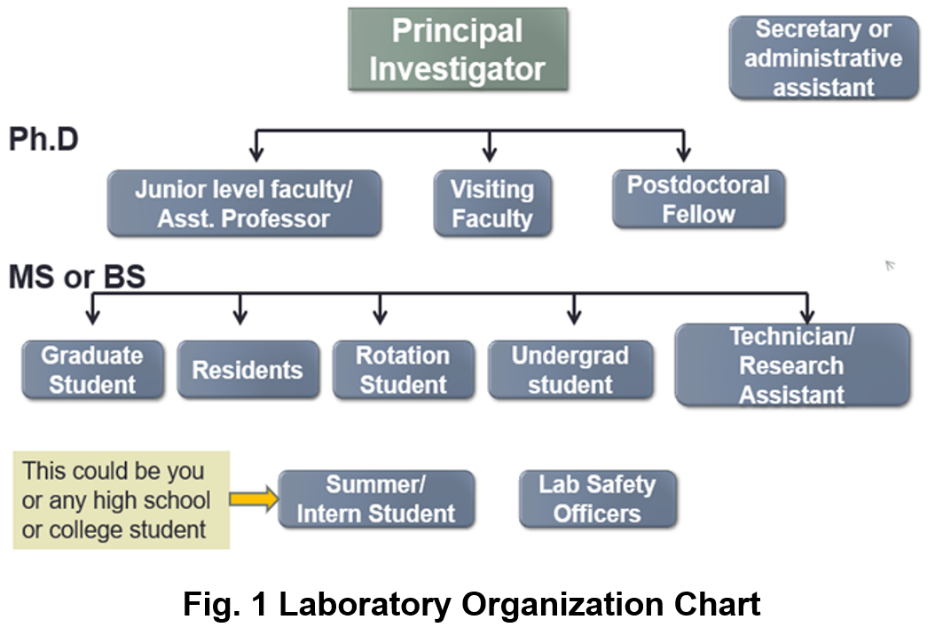
Graduate students are engaged in research on their PhD project(s). The lab can also have medical residents, undergraduate students, or research technicians. Many labs also accept summer students or internship students for a brief period of time to allow them to have research experience. The lab personnel can have varying educational background, such as high school diploma, college certificate, associate, bachelor’s degree or higher.
Research Labs in Companies: Biotech Companies provide products and/or services utilizing biotechnology. The products produced can vary from food supplement to medications. Cetus, the first biotech company, was established in 1971 by Nobel L aureate Donald Gasler and others[1]. Cetus primarily developed microbial processes for producing chemical feedstocks such as propylene oxide and antibiotic intermediates. Another major biotechnology company called Genentech was founded in 1976 by Dr. Herbert W Boyer and Robert A Swanson. It was also the first biotech company backed by funding to explore the commercial potential of genetic engineering for drug discovery[2]. Genentech’s primary accomplishment was the production of human somatostatin in 1977. This was the first successful expression of human genes in bacteria. Genentech also made the first human synthetic insulin in bacteria in 1978. Before this, insulin was derived from the pancreas of cows and pigs. It is astonishing to learn that two tons of pig parts were needed to extract just eight ounces of insulin. Thus, the synthetic and large scale production of insulin helped meet supply demands. Genentech is a part of Roche since 2009. Today there are thousands of biotech companies in US that produce drugs, pharmaceutics and products for human health. Table 1 shows the top 10 Biopharma Clusters[3] and Biotech Companies[4] in USA.
Table 1 : Top 10 Biopharma Clusters and Biotech Companies in USA
Top 10 BioPharma Clusters
| Top 10 Biotech Companies
|
Maryland, Washington DC and Virginia comprise the BioHealth Capital Region (BHCR). There are 1,800 life sciences companies, over 70 federal labs and world class academic and research institution[5]. BHCR and is considered as the fourth largest Biopharma cluster in the nation. Hence there is a huge need of workforce at various levels that can help these companies and labs thrive.
Biotechnician or a laboratory/ manufacturing technician must have knowledge and skills so they are able to assist and perform biological experiments and processes in biotech or pharmaceutical industry they work in. Their work can often involves making buffers, solutions, and media. They must be able to always follow directions accurately. To perform any procedure in a biotech company, technicians are trained and able to follow Standard Operating Procedures (SOPs), which means they must follow always the exact same steps to complete a procedure. They must be completely knowledgeable of practices and procedures that prevent contamination from pathogens (aseptic techniques) and take great care to keep the work area free of any contamination. They should be able to follow Good Laboratory Practices (the regulations that help laboratories operate). Technicians should also be able to perform experiments, analyze data, produce graphs and maintain appropriate laboratory notebook or records as required.
Core Labs: Core Lab supports biological, clinical, and animal research by providing cost-effective laboratory testing or analysis. Typically core labs have state-of-the-art technology not easily affordable by every lab, so they provide the service and expertise to all in in universities, research institutes, and commercial laboratories. Core facilities might specialize in technology such as DNA (Deoxyribonucleic acid) sequencing, imaging, bioinformatics, cell/ tissue culture and flow cytometry, lab animal services, biorepository, pathology/ histology, pharmaceutical and drug discovery and more. The focus of core lab is to provide services which is very specialized so that the researchers do not have to become expert in all the areas. The technicians working in core laboratories should have the basic skills and the aptitude and intent to learn the particular skill in the core lab that they want to work in. They also might need to work with different people every day and should be capable of good communication skills.
Some examples of a core lab are as follows:
- Bioinformatics: Help researchers understand large and/or complex data sets using computer analysis and statistics.
- Flow Cytometry: Flow cytometry is a method in which researchers sort cells into groups by labeling them with different colored fluorescent labels. Personnel specialize in the use of flow cytometry equipment and analysis software.
- Lab Animal Services: Personnel in this area care for animals being used for research; monitor for proper care and treatment; maintain approved protocols and ensure appropriate use of animals per protocols.
- Mass Spectrometry: provides a wide variety of chemical analyses using mass spectrometry techniques for organic and biological samples.
- Nuclear Magnetic Resonance (NMR): NMR is a series of techniques to determine chemical structures. Researchers in this lab specialize in the use of the equipment and software related to NMR.
- Nuclear Acid and Protein Research: Scientists in these labs are experts in techniques related to isolating and analyzing nucleic acids (DNA, RNA) and proteins
- Pathology: Personnel in this lab prepare, stain and examine tissues microscopically, including using various types of microscopes, web‐based data applications, and 3‐D imaging.
Clinical Labs: A medical or clinical laboratory is a place where tests are usually done on clinical specimens in order to obtain information about the health of a patient as pertaining to the diagnosis, treatment, and prevention of disease. Research in clinical labs deal primarily with the clinical samples. Clinical laboratory staff are responsible for conducting studies to examine the safety and efficacy of new drugs in humans. This job involves performing routine sample analysis from patients, maintaining, and reviewing documents to ensure that they are in compliance with protocols, regulatory requirements, and SOPs (standard operating procedures). Here are some examples of clinical laboratories and the role of lab personnel:
- Blood bank: Personnel in this lab specialize in delivery and appropriate treatment with blood and blood products.
- Microbiology: Personnel in this lab analyze samples for pathogens such as bacteria, yeast.
- Immunogenetics: Personnel in this lab perform tests associated with HLA‐typing, a kind of genetic test to increase successful outcomes following transplants and for diagnosis of diseases.
- Molecular Genetics: Personnel in this lab perform assays that help with diagnosis and treatment of genetic disorders, such as clotting disorders and cancer predisposition syndromes.
- Stem Cell Lab: Personnel in this lab prepare progenitor/stem cell populations for transplantation, process and store donor samples, and perform assays to enrich sub‐populations of cells within the sample prior to transplant.
Clinical trials are experiments that involves determining outcomes of a new treatment such as vaccine, drug, dietary supplement and medical devices in human participants. Patient research in clinical labs is divided into 4 phases[6].
- Phase I trials: There are often conducted in 20-100 (small number) of healthy volunteers or people with disease/ condition. They involve first-in-human evaluation to evaluate safety, tolerability, and determine appropriate dosage. About 70% of the drugs move to the next phase.
- Phase II trials: Intervention given to a larger group (100-300) with disease/ condition to evaluate effectiveness and safety. About 33% of the drugs move to the next phase.
- Phase III trials: Intervention given to large groups (upto thousands) to confirm effectiveness, monitor side effects, compare to other treatments, and collect information that will allow it to be used safely. About 25-30% drugs move to the next phase.
- Phase IV trials: Thousands of volunteers may participate in the study to determine safety and efficacy of the drug. Post marketing studies determine additional information including risks, benefits, and optimal use of an intervention.
To get a full spectrum of idea of the numerous lab technician jobs, their roles and the salaries, take a look at the job descriptions at www.biotech-careers.org or click on the specific type of role(s) in table 2 that incite your interest.
Table 2: Laboratory Technician jobs in various fields
In summary, the various types of research, basic, translational and clinical research are interdependent as shown in figure 3. They are vital for our understanding of scientific processes as well as for the improved health of the patients.
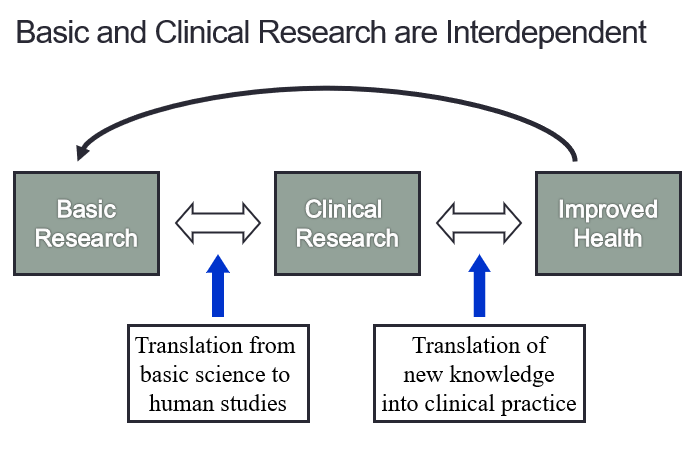
Fig 3. Correlation of Basic & Clinical Research with human health
1.2 Introduction to Laboratory Quality Management System
A clinical or medical laboratory is a laboratory where tests are carried out on clinical or patient specimens to obtain information about the health of an individual. This can aid in diagnosis, treatment, and prevention of disease. At any given point of time these labs may have hundreds or thousands of samples undergoing numerous laboratory tests. At the core of this process, samples are collected, received, registered, and then processed. The concept of quality management will be explained in context of medical laboratories.
Some of the important features of medical laboratories are:
- Highly complex operations
- Individuals doing complex tasks
- Absolute need for Accuracy
- Absolute need for Confidentiality
- Absolute need for Time Effectiveness
- Absolute need for Cost Effectiveness
Seventy percent of clinical medicine decision making is predicated upon or confirmed by medical laboratory test results. In the United States, there are between 7-10 billion laboratory tests reported annually. In a 5 country study, 15% of patients were reported to receive either incorrect or delayed reports on abnormal results[7]. Hence there is an continuous and absolute necessity for quality management.
Quality can be defined as conformance to requirements of a product. In laboratory, quality can be defined as accuracy, reliability, and timeliness of reported test results. The laboratory results must be as accurate as possible, all aspects of the laboratory operations must be reliable, and reporting must be done in timely manner for it to be useful in a clinical or public health setting.
Quality management system can be defined as “coordinated activities to direct and control an organization with regard to quality”[8].
This definition is used by internationally recognized laboratory standards organizations such as International Organization for Standardization (ISO)[9] and Clinical and Laboratory Standards Institute (CLSI)[10].
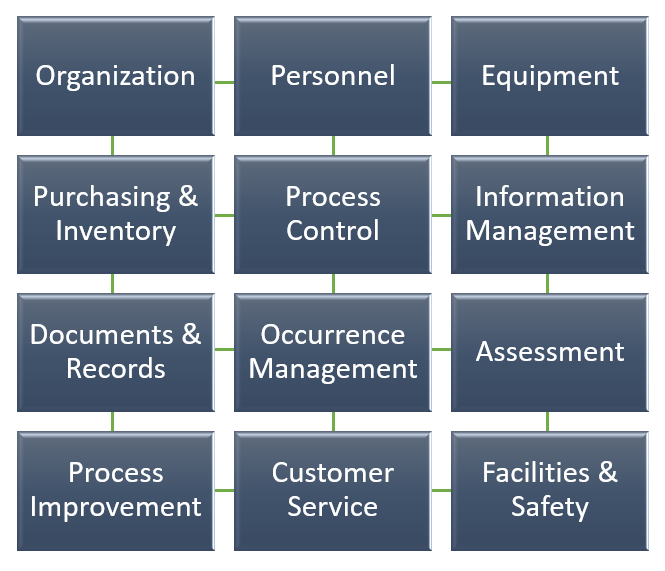
A quality management system can be described as a set of building blocks needed to control, assure and manage the quality of the laboratory's processes. A system used in this tool is the framework of 12 building blocks, called quality system essentials (QSEs). These quality system essentials are a set of coordinated activities that serve as building blocks for quality management[11].
|
Good quality in processes, equipment and performance is indispensable for the overall good performance of the laboratory.
1. Organization
Aims to create a functioning quality management system
- The structure and management of the laboratory must be organized
- Quality policies can be established and implemented.
- There must be a strong supporting organizational structure with commitment from the management to ensure a functioning quality management system.
- There must be a mechanism for implementation and monitoring
2. Personnel
The most important laboratory resource is competent and motivated staff.
- The quality management system addresses many elements of personnel management and oversight.
- The importance of encouragement and motivation
3. Equipment
Each piece of equipment or instrument used in a lab must be calibrated and functioning properly.
- The right equipment must be chosen and installed correctly.
- The new equipment should be tested to ensure that its functioning properly .
- There should be a system for maintenance of equipment.
4. Purchase and Inventory
Proper management of purchases and stock/ inventory are important for cost savings.
- Supplies and reagents must be available when needed
- The procedures that are a part of management of purchasing and inventory must be designed to ensure that all reagents and supplies are of good quality.
- The reagents and supplies must be used and stored in a manner that preserves integrity and reliability.
5. Process Control
The laboratory must institute process control to ensure the quality of the laboratory testing processes.
- Process control factors include:
- Quality control for testing,
- Appropriate management of the sample, including collection and handling
- Method verification and validation.
- The elements of process control are very familiar to laboratories; quality control was one of the first quality practices to be used in the laboratory and continues to play a vital role in ensuring accuracy of testing.
6. Information Management
The product of the laboratory is information, primarily in the form of test reporting.
- Information (data) must be carefully managed to ensure accuracy and confidentiality.
- It should be accessible to the laboratory staff and to the health care providers.
- Information may be managed and conveyed with either paper systems or with computers.
7. Documents & Records
Documents are needed in the laboratory to inform how to do things. Laboratories always have many documents.
- Records must be meticulously maintained so as to be accurate and accessible.
- Many of the 12 quality system essentials overlap.
- A good example is the close relationship between "Documents and records" and "Information management".
8. Occurrence Management
An “occurrence” is an error or an event that should not have happened.
- A system must be instituted to detect these problems or occurrences.
- The system must include steps to handle the “occurrence” properly, and include provisions to learn from mistakes
- Take action so that they do not happen again.
9. Assessment
The process of assessment is a tool for examining laboratory performance and comparing it to standards, benchmarks or the performance of other laboratories.
- Assessment may be:
- Internal (performed within the laboratory using its own staff)
- External (conducted by a group or agency outside the laboratory).
- Laboratory quality standards are an important part of the assessment process, serving as benchmarks for the laboratory.
10. Process Improvement
The primary goal in a quality management system is continuous improvement of the laboratory processes.
- This must be done in a systematic manner.
- There are a number of tools that are useful for process improvement.
11. Customer Service
Customer service is essential for medical laboratory as it is a service organization.
- It is essential that clients of the laboratory receive what they need.
- The laboratory should understand who the customers are, and should assess their needs and use customer feedback for making improvements.
12. Facilities & Safety
Many factors must be a part of the quality management of facilities and safety. These include:
- Security—which is the process of preventing unwanted risks and hazards from entering the laboratory space.
- Containment—which seeks to minimize risks and prevent hazards from leaving the laboratory space and causing harm to the community.
- Safety—which includes policies and procedures to prevent harm to workers, visitors and the community.
- Ergonomics—which addresses facility and equipment adaptation to allow safe and healthy working conditions at the laboratory site.
In the quality management system model, all 12 quality system essentials must be addressed to ensure accurate, reliable and timely laboratory results. It is important to note that the 12 quality system essentials may be implemented in the order that best suits the laboratory. Approaches to implementation will vary with the local situation.
Laboratories that do not implement a good quality management system will result in many errors and problems that could even go undetected. Implementing a quality management system may not guarantee an error-free laboratory, but it does yield a high-quality laboratory that detects errors and prevents them from recurring.
[1] First-Hand: Starting Up Cetus, the First Biotechnology Company - 1973 to 1982 - Engineering and Technology
History Wiki. www.ethw.org
[2] Russo, E. (2003). "Special Report: The birth of biotechnology". Nature. 421 (6921): 456–457. Bibcode:2003Natur.421..456R. doi:10.1038/nj6921-456a. PMID 12540923. S2CID 4357773.
[3] https://sciencecenter.org/news/top-10-u-s-biopharma-clusters
[4] https://en.wikipedia.org/wiki/List_of_largest_biotechnology_and_pharmaceutical_companies
[5] http://www.biohealthcapital.com
[6][6] https://www.fda.gov/patients/drug-development-process/step-3-clinical-research
[7] Boone DJ, IQLM, 2005
[8] https://www.iso.org/iso-9001-quality-management.html
[9] www.iso.or
[1] Merriam-Webster (n.d.). In Merriam-Webster.com dictionary. Retrieved February 13, 2021, from https://www.merriam-webster.com/dictionary/laboratory.
Quality Control, Assurance & Management
Where do you hear the word ‘quality’ in daily life? When you call a company's customer service line, you often hear the words "this call may be monitored or recorded for quality assurance purposes," at the beginning of the conversation. Quality is of utmost importance for every organization that serves people in any way.
The word has quality originates from Latin word qualitas. Qualitas means “general excellence” or “a distinctive feature”[1]. Good quality is like reputation, it takes a long time to build but can be ruined with one mistake. If you set 99% as a level of quality it means that you are willing to accept a 1% error rate.
One percent error rate would mean: 5 bad landings/ take offs out of 500 flights every day at a medium sized airport. It could mean 0.2 million out of 20 million packages lost or delivered to wrong address around US every day by USPS. None of these are acceptable outcomes, hence we know that highest standards of quality are needed in every aspect of life.
Quality is defined as the totality of features and characteristics of a product or service that bears on its ability to satisfy stated or implied needs (ISO 1994).
Quality varies from different viewpoints:
- User-based: better performance, more features
- Manufacturing-based: conformance to standards, making it right the first time
- Product-based: specific and measurable attributes of the product
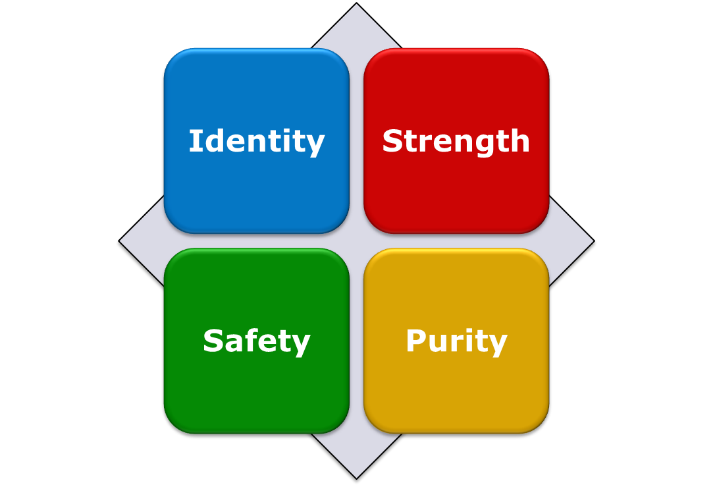
Every pharmaceutical product must meet four attributes namely: Identity, Strength, Safety and Purity[2].
Achieving quality means that these attributes are achieved for every product.
In health care system essentially, quality means laboratory results that are
- Accurate,
- Reliable,
- Timely
Quality in a complex laboratory system could be divided in three phases pre-analytic, analytic and post-analytic as shown below[3].
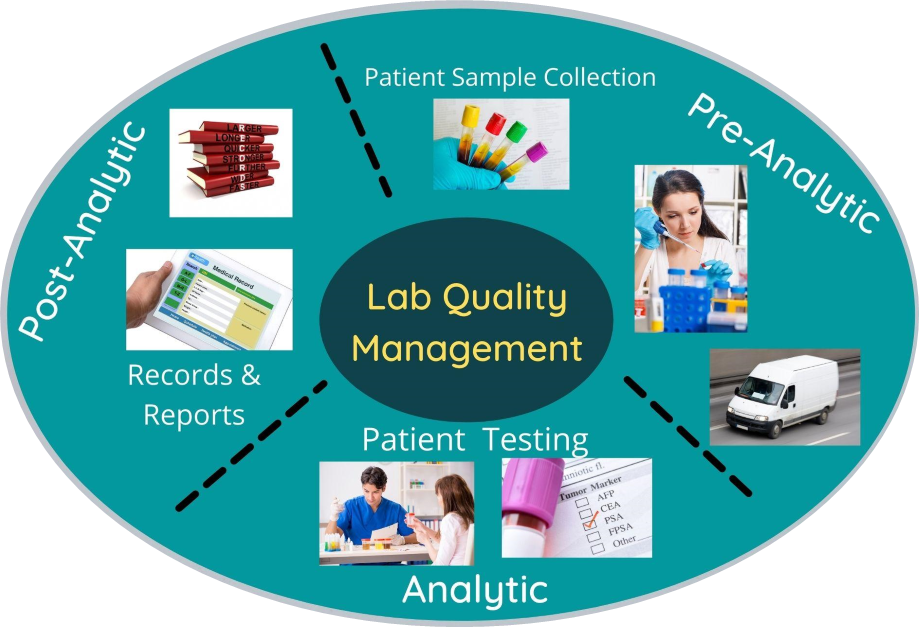
Fig. 6 Quality in a Complex Laboratory System
There are 4 types of complexities in the laboratory system:
- Mistakes & defects
- Breakdowns & delays
- Inefficiencies
- Variation
These laboratory errors can cost in 1) Time 2) Personnel effort and 3) Patient outcomes. A report published in 2016 attributed 250,000 deaths per year due to medical errors in the US. This is an average over an eight-year period. In a 2006 publication, it was mentioned that 17% of adult patients in the US experienced medical, medication or lab errors[4], [5].
So as to achieve ‘quality’ or ‘excellent performance’ in the laboratory, according to the US FDA ‘Quality should be built into the product, and testing alone cannot be relied on to ensure product quality”
- Building quality into the product involves having controls at every stage of manufacturing and not only terminal controls.
- These include controls on all input resources like people, facilities, equipment, materials, process and testing etc.
Following variables may affect ultimate quality of product
- Raw material
- In process variations
- Packaging material
- Labeling
- Finish product
- Manual Error
All aspects of the laboratory operation are needed to be addressed to assure quality, this constitutes a quality management system. Quality management system (QMS) is the coordinated activities to direct and control an organization with regards to quality.
The 12 Quality Management System (QMS) essentials that you read in Chapter 1 are an integral part of healthcare and help in achieving high quality standards. They can be divided in three parts for a laboratory quality management systems purpose.
Table 3: QMS essentials categorized into Laboratory, Work and Outome
Laboratory | Work | Outcome |
Organization Facilities & safety Personnel Equipment Purchasing & inventory | Process Control Documents & records Information management | Occurrence management Assessments Customer service Process improvement |
Pareto Principle in Quality Management
One of the important tools for Quality Management is Pareto chart based on the Pareto principal. Vilfredo Pareto, an Italian engineer and economist, first observed the 80/20 rule in relation to population and wealth. He noted that in Italy and several other European countries, 80% of the wealth was controlled by just 20% of the population. Pareto principal is the 80–20 rule or the predication that for many events, roughly 80% of the effects come from 20% of the causes.
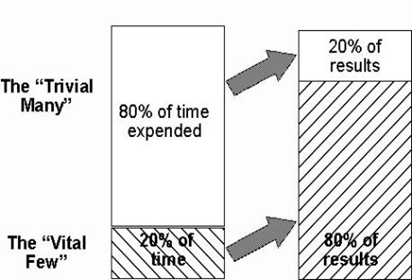
Fig. 7. Pareto Principle – Time vs Results
Quality management pioneer Dr. Juran implemented Pareto’s principle to Quality management. In simple words, we spend 80% of time doing 20% of tasks, and we spend only 20% time doing 80% of tasks that matter most. In a manufacturing process, 80% of the downtime might result from 20% of the problems. Pareto chart is a bar chart showing how much each cause contributes to an outcome or effect[1], [2].
Total Quality Management (TQM) approach of an organization is centered on quality, based on the participation of all its members and aiming at long term success through customer satisfaction and benefits to all members of the organization and society. – ISO (International Organization of Standardization).
Total Quality Management (TQM) involves quality management of company, processes and products. Quality control is a part of quality assurance and quality assurance is a part of quality management.
- TQM involves coordinated and customized management effort with focus on quality.
- QA is responsible for ensuring quality requirements are being met.
- QC tests & maintains established quality criteria for products.
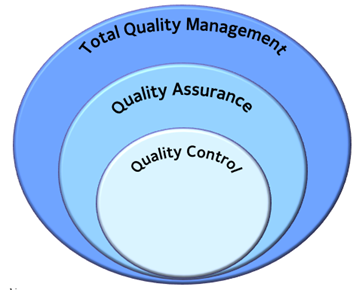
Fig. 8. Relationship – QC, QA and QM
Table 4: Differences between QA & QC
Basic Differences between QA & QC | |
QA approves test, method, standards & ensure that high standard must confirm with Good Manufacturing Practices & with QC facility. | QC is one compartment of multicompartment QA system. |
QA is interested in what happened yesterday, what is happening today, what is going to happen tomorrow. | QC is interested in what is happening today. |
Definition | |
QA is a set of activities for ensuring quality in the processes by which products are developed. | QC is a set of activities for ensuring quality in products. The activities focus on identifying defects in the actual products produced. |
QA is a managerial tool | QC is a corrective tool |
Goals & Focus | |
QA aims to prevent defects with a focus on the process used to make the product. It is a proactive quality process. | QC aims to identify (and correct) defects in the finished product. Quality control, therefore, is a reactive process. |
The goal of QA is to improve development & test processes so that defects do not arise when product is being developed. | The goal of QC is to identify defects after a product is developed and before it's released. |
What & how does it work? | |
QA works by prevention of quality problems through planned and systematic activities including documentation. | QC works by the activities or techniques used to achieve and maintain the product quality, process and service |
QA establishes a good quality management system and the assessment of its adequacy. | QC finds & eliminates sources of quality problems through tools & equipment so that customer's requirements are continually met. |
Whose responsibility is it & example? | |
Everyone on the team involved in developing the product is responsible for quality assurance. | Quality control is usually the responsibility of a specific team that tests the product for defects. |
Verification is an example of QA. | Validation is an example of QC. |
Quality Assurance Unit comprises of personnel or organizational element, designated by management to perform the duties relating to QA of nonclinical laboratory studies.
Responsibilities of QA department:
- The QA department is responsible for ensuring that the quality policies adopted by a company are followed.
- It must determine that the product meets all the applicable specifications and that it was manufactured according to the internal standards of GMP.
QA also holds responsible for quality monitoring or audit function.
The concept of total quality control refers to the process of striving to produce a perfect product by a series of measures requiring an organized effort at every stage in production. Although the responsibility for assuring product quality belongs principally to QA personnel, it involves many departments and disciplines within a company. To be effective, it must be supported by team effort. Quality must be built into a drug product during product and process, and it is influenced by the physical plant design, space, ventilation, cleanliness and sanitation during routine production.
Advantages of TQM: The advantages of TQM are 1) Improves reputation - faults and problems are spotted and sorted quicker. 2) Higher employee morale - workers motivated by extra responsibility, teamwork and involvement in decisions of TQM. 3) Lower cost - decrease waste as fewer defective products and no need for separate. 4) Quality control inspector.
Disadvantages of TQM: The disadvantages of TQM are 1) Initial introduction cost. 2) Benefits of TQM may not be seen for several years. 3) Workers may be resistant to change.
QMS Essentials model and TQM provides the roadmap that can successfully provide the laboratory’s best contribution to patient care and ensure safety and quality control of products in the pharmaceutical industry.
[1] Pareto, Vilfredo; Page, Alfred N. (1971), Translation of Manuale di economia politica ("Manual of political economy"), A.M. Kelley, ISBN 978-0-678-00881-2
[2] Code of Federal Regulations, Title 21, https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211&showFR=1
[3] Laboratory Quality Management System Handbook. https://www.who.int/ihr/publications/lqms/en/
[4] Health for a life. https://www.aha.org/system/files/content/00-10/071204_H4L_HighestQualityCare.pdf
[5] Makary Martin A, Daniel Michael. Medical error—the third leading cause of death in the US BMJ 2016; 353 :i2139
[6] Pareto, Vilfredo; Page, Alfred N. (1971), Translation of Manuale di economia politica ("Manual of political economy"), A.M. Kelley, ISBN 978-0-678-00881-2
Quality Management Regulation – US Regulatory Agencies
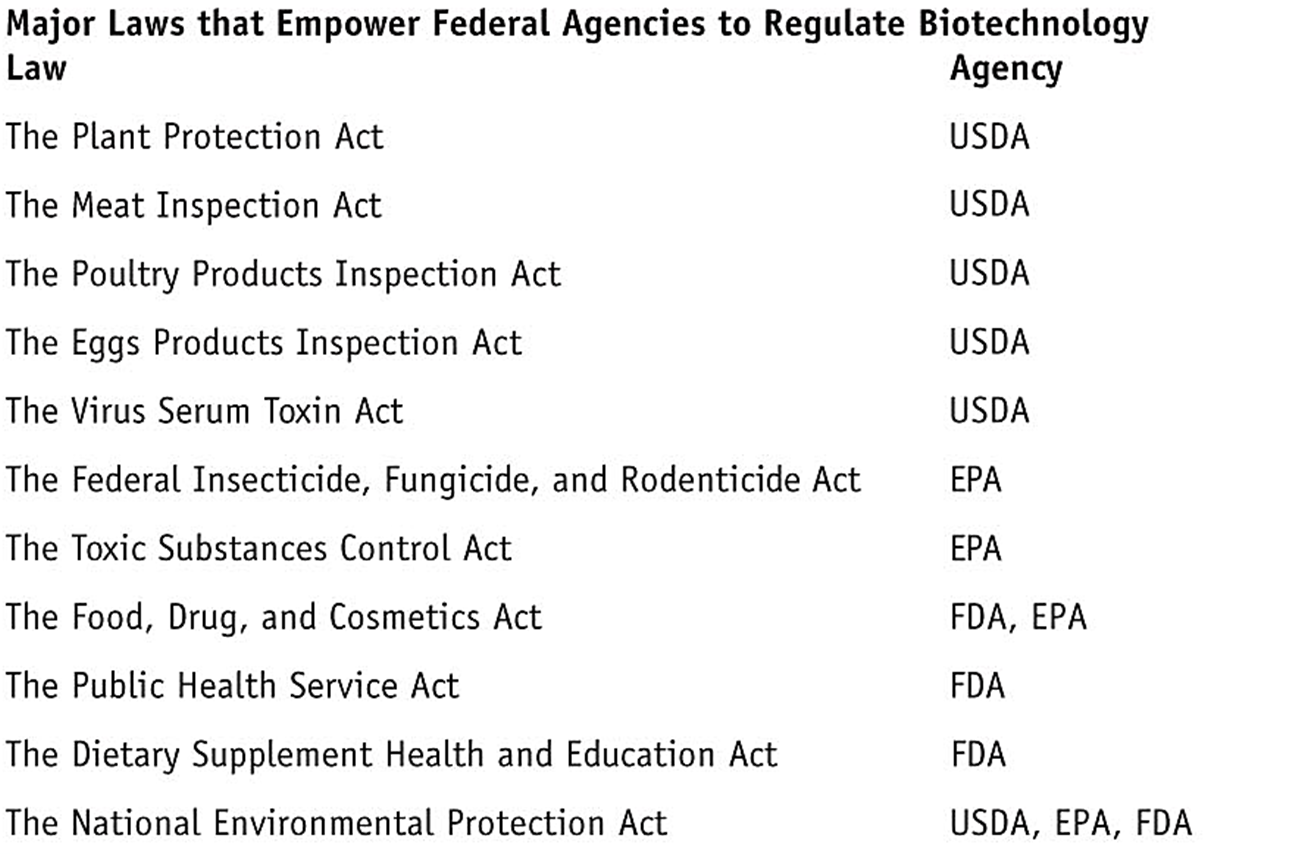
We have learned that the QMS ensures that the product meets quality standards set by local and national governing bodies. QMS helps bring products to market that meet the requirements of regulatory agencies and customers. Different regulatory agencies are responsible for different products. For example, US Department of Agriculture (USDA) regulates plants, plant-pests, animal vaccines; Environmental Protection Agency (EPA) regulates microbial/ plant pesticides, toxic substances, microorganisms, animal producing toxic substances; Food & Drug Administration (FDA) regulates food additives, human & animal drugs, human vaccines, medical devices, transgenic animals, and cosmetics. These regulatory agencies have laws that empower them to regulate biotechnology products in the market. Some of the laws and respective regulatory agencies are listed here.
Our focus in this chapter will be on the FDA.
FDA’s Role and Responsibilities:
- FDA regulates drugs, foods, cosmetics, biologics, medical devices.
- FDA is responsible for administration, enforcement, interpretation of US drug law and has power to establish regulations which have the force and effect of law
- FDA has developed policies, procedures, and regulations to implement its Regulatory initiatives.
History of drugs and regulations
In 1848, most of the drugs were imported in the USA . Unregulated production of tetanus antitoxin. In 1902, unregulated productions of tetanus antitoxin resulted in illness and death. On October 26, 1901, a five-year-old St. Louis girl died from tetanus. 13 more children in St. Louis died of tetanus, and the cause was traced to a supply of diphtheria antitoxin prepared by the St. Louis Board of Health from a tetanus-infected horse. The St. Louis disaster wasn't the only such incident in the United States and Europe. Camden, New Jersey, was the site of almost a hundred cases of post-vaccination tetanus, including the deaths of nine children, in the fall of 1901.

1902: Biologics Control Act was passed on July 1, 1902. Biologics had to be labeled with the name and license number of the manufacturer, and the production had to be supervised by a qualified scientist. Establishments were required to have annual licensing.
The Hygienic Laboratory, forerunner of the National Institutes of Health, was authorized to conduct regular inspections of the establishments and to sample products on the open market for purity and potency testing. Later in 1972 FDA was handed over this role.
1906: Harvey Wiley, head of the Bureau of Chemistry of the U. S. Department of Agriculture, led the way toward consumer protection by working towards a law, Pure Food and Drugs Act.

The Food & Drugs Act was not completely devoid of problems, it resulted in the Sulfanilamide tragedy. The 1906 act failed to regulate medical devices or cosmetics, the lack of explicit authority to conduct factory inspections, the difficulty in prosecuting false therapeutic claims following a 1911 Supreme Court ruling, and the inability to control what drugs could be marketed.

1938: The Elixir Sulfanilamide disaster reinvigorated a bill to replace the 1906 act.
Roosevelt signed the Food, Drug, and Cosmetic Act into law on June 25, 1938. The 1938 act required that firms had to prove to FDA that any new drug was safe before it could be marketed--the birth of the new drug application. The new law covered cosmetics and medical devices, authorized factory inspections, and outlawed bogus therapeutic claims for drugs. Drugs had to bear adequate directions for safe use, which included warnings whenever necessary.

1951: The 1938 act was vague on issues such as what a prescription was and who would be responsible for identifying prescription versus non-prescription drugs. This lack of statutory direction created many battles between FDA, regulated industry, and professional pharmacy, and within some of these groups as well. The 1951 Durham-Humphrey Amendment to the 1938 act helped clarify some of these disputed issues.


1962: Kefauver Harris amendment was introduced. The amendment was a response to the Thalidomide tragedy, in which thousands of children in Europe were born with birth defects as a result of their mothers taking thalidomide for morning sickness during pregnancy. Thalidomide had not been approved for use in the United States. The bill by U.S. Senator Estes Kefauver, of Tennessee, and U.S. Representative Oren Harris, of Arkansas, required drug manufacturers to provide proof of the effectiveness and safety of their drugs before approval.

1983: Orphan diseases are serious and debilitating rare diseases affecting less than 200,000 people, which typically receive little funding toward their prevention or treatment. About 20 million Americans suffer from at least one of the more than 5000 known rare diseases.
Representative Henry Waxman of California initiated hearings into the lack of drugs for orphan diseases. The Orphan Drug Act finally became law in 1983.

1990s: By 1990s, Standard drug testing and approval process was in place. This included:
- Preclinical testing
- Clinical studies – Phase I, II, III
- Pre Market Analysis (PMA)
- Post marketing surveillance
The Prescription Drug User Fee Act (PDUFA) of 1992 authorized FDA to charge user fees for certain drugs and biological product applications. These user fees support timely reviews for drugs. PDUFA has enabled the FDA to bring access to new drugs fast while maintaining the same thorough review process. PDUFA may not need to be paid for orphan drugs as exception.
Since PDUDA was passed, more than 1000 drugs and biologics have come to the market including new medicines to treat cancer, AIDS, cardiovascular disease and life-threatening infections.
Current Role of FDA in Drug Development
The FDA’s role is to evaluate data in an investigational drug and determine if it should be made available to the patients. The benefits of the proposed drug must outweigh risks and side effects. The FDA also issues directive on the right dosage and information on how to use the medication properly.
The FDA outlines its drug approval process as a 12 step process that includes 4 phases. The first step involves animal testing. If the drug proves to be non-toxic to animals, then the sponsor can submit an IND (Investigative New Drug Application), after which the drug goes into clinical trials and or studies. The clinical trials are separated into three initial phases.
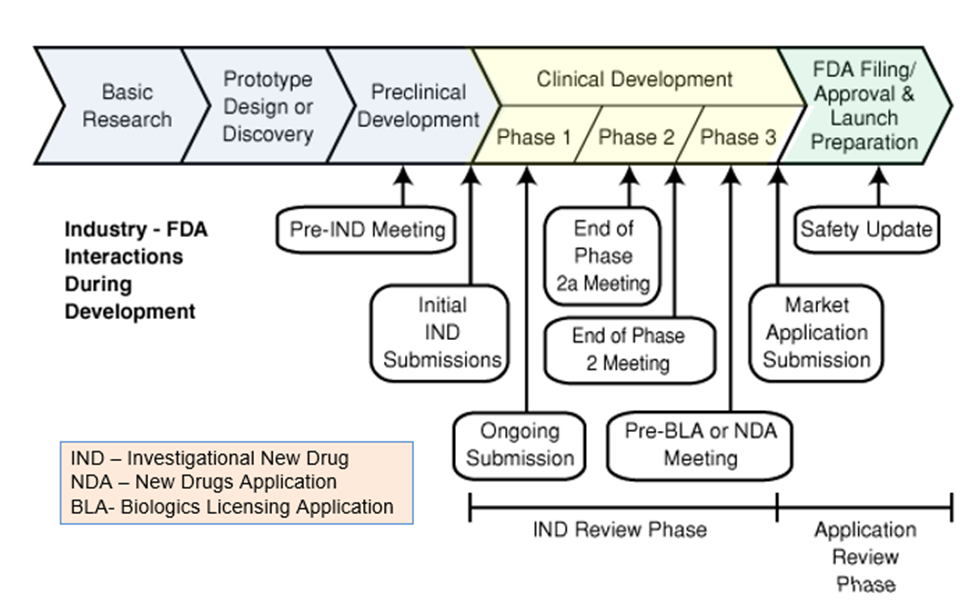
In case of emergencies such as the COVID-19 pandemic, FDA has created the Coronavirus Treatment Acceleration Program (CTAP). This program is responsible for managing the delicate balance between expediting the process of getting much-needed therapies approved for public use while simultaneously monitoring the safety of said therapies.
Despite various reforms to FDA’s processes, developing new medicines has become increasingly expensive and time-consuming. The Critical Path Initiative is FDA’s effort to address the need for up-to-date scientific means of evaluating the safety, efficacy, and quality of medicines. The object of this initiative is to reduce the time, cost, and uncertainty of product development.
The Critical Path Initiative
° Creates new scientific tools for safety, efficacy, and quality
° Strives to reduce time, cost, and uncertainty
Regulatory Framework
Regulatory framework comprises of three parts:

FDA’s Quality Systems Inspection Techniques
The Quality System Inspection Technique, or QSIT, is a plan for investigators to follow when evaluating a manufacturer’s compliance with regulations for Quality Systems, Medical Device Reporting, Corrections and Removals, and Tracking.
The QSIT handbook also includes guidance for covering sterilization when the Production and Process Control subsystem is inspected.
FDA has identified 7 subsystems in the Quality System.
- You can think of these subsystems as types of activities.
- You can find these subsystems identified clearly in the Quality System Regulation.
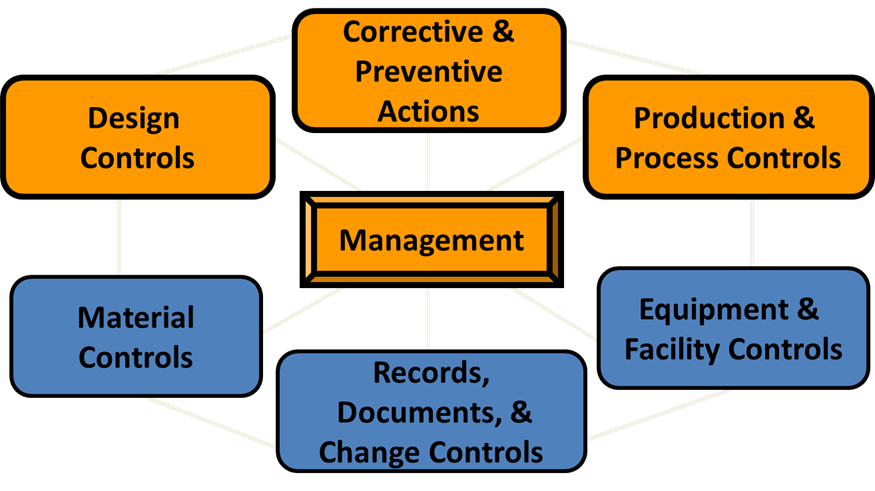
The four subsystems in focus are:
- Management Control
- Design control
- Corrective & Preventive Actions
- Production & Process Control
Management Control:
The purpose of this subsystem is to provide 1) adequate resources, 2) ensure the establishment and effective functioning of the quality system, and 3) monitor the quality system and make necessary adjustments.
Management Subsystem has the following requirements:
- Conduct management reviews
- Appoint Management representative and required personnel
- Establish Quality Policy along with objectives & organizational structure.
- Establish Quality audit procedures and quality audits
Design Controls:
The purpose of this subsystem is to provide 1) Control of the design process 2) Assure the device design meets used needs, intended uses and the specified requirements.
Design Controls subsystem has the following requirements:
- Establish a plan that describe or reference design and development activities
- Identify design inputs
- Develop design outputs
- Verify that design outputs meets design inputs
- Validate the design (include software validation and risk analysis)
- Control design changes
- Review design results
- Transfer the design to production
- Compile a design history file
Corrective & Preventive Action (CAPA)
The purpose of this subsystem is to 1) Collect and analyze information/data 2) Identify and investigate product and quality problems 3) Identify, implement and validate effective corrective and preventive action 4) Communicate CAPA to appropriate personnel.
CAPA has the following requirements:
- CAPA procedures must be established.
- Investigations must be conducted to identify root cause of failures.
- Appropriate corrective actions and preventive actions must be carried out.
- Personnel must be trained on CAPA activities
- Management must review of CAPA activities
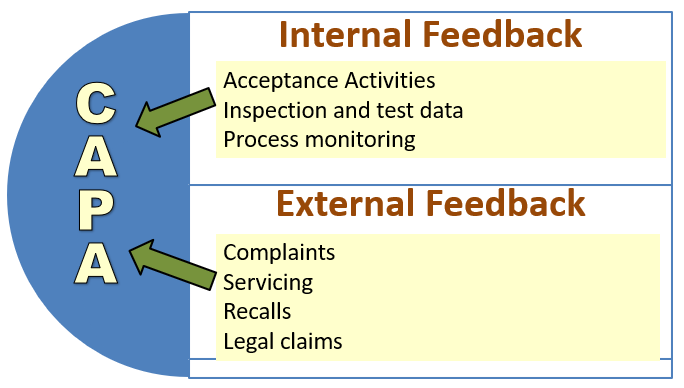
Post-Inspection:
The company where the audit is completed, will send a letter to FDA identifying how they have corrected deficiencies or will correct them. They may also provide documentation of any corrections that have been completed. They may also provide a timetable or estimated completion date for future corrections
Types of warning letter
If a company/ manufacturer has significantly violated FDA regulations, FDA notifies the manufacturer in the form of a Warning Letter. The Warning Letter identifies the violation, such as poor manufacturing practices, problems with claims for what a product can do, or incorrect directions for use. There are different types of warning letters
- General FDA Warning Letters
- Tobacco Retailer Warning Letters
- Drug Marketing and Advertising Warning Letters (and Untitled Letters to Pharmaceutical Companies)
The Office of Prescription Drug Promotion (OPDP) regulates prescription drug promotional materials made by or on behalf of the drug’s manufacturer, packer, or distributor, including: 1) TV and radio commercials 2) Sales aids, journal ads, and patient brochures 3) Drug websites, e-details, webinars, and email alert.
FDA’s Bad Ad Program
This program was launched in 2010. It was designed to educate healthcare providers about the role they can play in helping make sure that prescription drug advertising is truthful. It provides an easy way to report misleading information to the agency (E-mail BadAd@fda.gov or call 855-RX-BADAD or by going to Fda.gov/badad)
Good Laboratory Practices
In early 1900s, a number of acts began the regulatory process in USA. FDA was recognized by its name in 1930s but its function had begun much earlier :

1947: Nuremberg Code was drafted in 1947. This was a 10 point code that described the basic principles of ethical behavior in the conduct of human experimentation
Three pivotal points were stressed:
- 1. Voluntary consent of the subject must be obtained.
- 2. Prior animal experimentation to determine risk must be performed.
- 3. Human experimentation must be performed by qualified medical personnel
1958- Food Additives Amendment came into effect in 1958. This act enforced the following:
- Required manufacturers of new food additives to establish safety
- Prohibited the approval of any food additive shown to induce cancer in humans or animals
1960- Color Additive Amendment passed in 1960 enforced the following measures of food safety:
- Required manufacturers to establish safety of color additives in foods, drugs, and cosmetics.
- Prohibited approval of color additives shown to induce cancer in humans/ animals.
1962- Thalidomide Tragedy
In 1962, a new sleeping pill caused birth defects in thousands of babies in Europe. Dr. Frances Kelsey refused to allow the new drug to become effective in the US because of insufficient safety data and saved thousands of life. In 1963, overseas thalidomide catastrophe was instrumental in the FDA completing the first draft of Good Manufacturing Practices (GMPs) and making them legal requirements. GMPs set requirements for sanitation, inspection of materials and finished products, record-keeping, and other quality controls.
1972- The Tuskegee Syphilis Experiment was an infamous clinical study conducted between 1932 and 1972 by the U.S. Public Health Service to study the natural progression of untreated syphilis in rural African-American men. They were told that they were receiving free health care from the U.S. government. The 40-year study was controversial for many reasons related to ethical standards because researchers knowingly failed to treat patients appropriately after the 1940s validation of penicillin as an effective cure for the disease they were studying. Revelation in 1972 of study failures by a whistleblower led to major changes in U.S. law and regulation on the protection of participants in clinical studies.
1974- As a result of this the National Research Act was passed. A commission was set up to define ethical standards under which research in the United States was to be conducted. The Commission, created as a result of the National Research Act of 1974, was charged with identifying the basic ethical principles that should underlie the conduct of biomedical and behavioral research involving human subjects and developing guidelines to assure that such research is conducted in accordance with those principles. Informed by monthly discussions that spanned nearly four years and an intensive four days of deliberation in 1976, the Commission published the Belmont Report, which identifies basic ethical principles and guidelines that address ethical issues arising from the conduct of research with human subjects. It was written in response to the infamous Tuskegee Syphilis study in which African Americans with syphilis were lied & denied treatment for more than 40 years. It established federal law requiring the review of all clinical research by an Institutional Review Board (IRB).

Events at Industrial Biotest that led to US Good Laboratory Practices (GLP)
There were several events that led to the eventual establishment of Good Laboratory Practices. However, the most infamous was the research discrepancies noted at a leading contract research laboratory, Industrial Biotest Corporation (IBT). IBT was an American industrial product testing facility in the US. IBT conducted studies for the federal government, as well as for private companies. In 1970s, IBT performed 35% to 40% of all toxicology tests.
However, based on FDA and EPA investigations, numerous discrepancies were uncovered. 618 of 867 (71%) of studies audited by the FDA were invalidated for having "numerous discrepancies between the study conduct and data.“ Consequently, IBT was at the center of one of the most far-reaching scandals in modern science, as thousands of its studies were revealed through EPA and FDA investigations to be fraudulent or grossly inadequate.
An employee of Syntex Corp. notified the FDA of problems (Whistle blower, a person who exposes any kind of information or activity that is deemed illegal, dishonest, or not correct within an organization that is either private or public).
Naprosyn, an antiarthritic drug tested by IBT. Syntex employee expressed concern to IBT officials about submitted report. The file provided enough evidence to warrant an inspection of the facility by the FDA.
Irregularities in IBT's data were discovered in April 1976 by Adrian Gross, an investigator at the FDA, whose aide retrieved one of the laboratory's naproxen studies that had been conducted for Syntex, a pharmaceutical company. The FDA proceeded to probe IBT. During Gross' physical inspection of the laboratory, he gained access to the study's raw safety data and found frequent references to an unknown acronym, "TBD/TDA," which he said perplexed him until learning that it denoted a testing animal whose body had "too badly decomposed.
IBT had a building where all of the rodents were housed, it was nicknamed "The Swamp". The Swamp was a horrible place. Its automatic watering system had faulty nozzles, that sprayed a chilling mist over the animals. The water filled their food jars, and some subsequently drowned. Some of those that didn't drown, died of exposure. They were wet and cold and could not survive. There were routinely 80% mortality rates in chronic studies. Technicians later told the FDA that the animals would decompose so rapidly that the bodies would ooze through the cage bottom. There were other problems... rats would escape through wire cages that were bent. There was a wild breeding colony that lived on the floor of the animal rooms. These wild rats would chew the toes off of the caged rats. Technicians would use chloroform to slow the wild rodents down to try to catch them. The chloroform fumes would kill some of the caged study animals.
The section Head for Rat Toxicology submitted fraudulent mortality data. 1000 new mice ordered to replace those that had died during the study. Blood and urine analyses were fabricated at the end of a 2 year rat study. Management forged signatures on reports. Eventually, there were indictments on 8 counts for conducting and distributing false scientific data.
IBT had over 22,000 studies to their credit. IBT studies were the basis for safe product rating for hundreds of drugs and pesticides. Of 1205 key pesticide studies, only 214 (18%) of the studies were found to be valid after retesting. Of 867 agency audits, 618 (71%) of the studies were found to be invalid. IBT was found to be engaging in extensive scientific misconduct and fraud, which resulted in the indictment of its president and several top executives in 1981 and convictions in 1983.
Firms that used IBT to test their products could not assume that the scientific data and reports submitted to the agencies were accurate and complete. Data was found to have been inaccurately reported as protocols weren't followed. Technicians may not have known that there was a protocol. Techs were not trained to keep accurate records or to make careful observations. Management did not assure proper supervision or critical review of data. There was no documentation of the qualification of technicians or scientists. Thousands of critical research projects were performed by IBT for nearly every major American chemical and drug manufacturer.

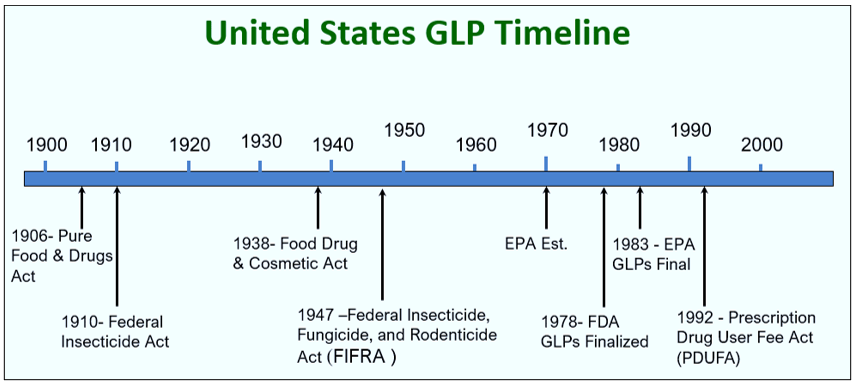
Problems at laboratories such as IBT, and many other laboratories prompted the need for a set of industry guidelines. Though intended to be guidelines, the problems with IBT forced the FDA to enact the Good Laboratory Practices (GLP) as law. This figure gives a basic timeline about the regulations and shows the development of the GLPs as we know of them today.
Good laboratory practice or GLP refers to a quality system of management controls for research laboratories and organizations to try to ensure the uniformity, consistency, reliability, reproducibility, quality, and integrity of chemical (including pharmaceuticals) non-clinical safety tests; from physio-chemical properties through acute to chronic toxicity tests.
GLP was first introduced in New Zealand and Denmark. It was proposed in USA in November 1976, implemented in June 1979, revised in Sep 1987 and minor changes were made to it in July 1991.
The United States FDA has rules for GLP in Title 21 is the portion of the Code of Federal Regulations (21 CFR 58) that governs food and drugs within the United States. Preclinical trials on animals in the United States of America uses these rules prior to clinical research in humans.

Scope of GLP:
GLPs apply to all studies supporting application/permits for FDA (Food & Drug Administration), EPA (Environmental Protection Agency), or international agencies.
GLPs do not apply to
- Basic exploratory studies
- Clinical studies (GCP)
- Testing in support of manufacturing (GMP)
GLP is a regulation that covers the quality management of non-clinical safety studies. The aim of GLP is to ensure that scientists organize and conduct the studies in a way that promotes the quality and validity of test data.

The purpose of GLP in scientific studies is
- To implement clear structures in the study.
- To ensure right procedures are being followed that are in compliance with GLP.
- To ensure that tests data and results are reliable.
- GLP is not involved in “Science” of the study but “Organization” of the study.
GLP does not aim to guide scientific studies or
- Tell what tests are to be performed.
- Tell what protocols are to be used.
- Assess the scientific value of the study.
These are achieved by scientific guidelines of the study not GLP.
GLP aims to make more obvious the incidence of FALSE NEGATIVES
(False negative : Results demonstrate non-toxicity of a toxic substance)
This doesn’t translate to incorrect clinical studies or harm to human but often results in waste of efforts and resources prior to clinical studies.
It also aims to make more obvious the incidence of FALSE POSITIVES
(False negative : Results demonstrate toxicity of a non-toxic substance)
This doesn’t translate to clinical studies as the compound may be valuable and useful but is discarded before clinical trials.
GLP aims to promote mutual recognition of studies across international frontiers.
30 countries are OECD members. If or when there was no GLP , many countries would refuse the outside drug or authenticity of studies and retrials had to be done.
OECD members and even non-OECD members now accept that studies have been conducted under acceptable organizational standards.
Essence and coverage of GLP:
GLP is a managerial concept for organization of the studies. This is particularly in reference to non-clinical studies. It includes dood & color additive petitions, NDA (New Drug Applications) & NADA (New Animal Drug Applications) and toxicity studies (in vitro & in vivo). It excludes human subject trials, clinical or field trials in animals, basic exploratory studies.
GLP defines the conditions under which studies are:
- Planned (Study plan/ protocol)
- Performed (Standard Operating Procedures)
- Recorded (Collection of raw data and deviations)
- Reported (The resulting final report has to be accurate)
- Archived (Study data, samples and specimens must be properly archived.)
- Monitored (Monitoring by study staff, Quality assurance personnel and quality inspectors)
The purpose of GLPs is to assure the quality & integrity of data submitted to FDA in support of the safety of regulated products. GLPs have heavy emphasis on data recording, record & specimen retention. The equipment should meet the following requirement.
- Equipment shall be adequately inspected, cleaned & maintained
- Equipment used for assessment of data shall be tested, calibrated and/or standardized
- Scales & balances should be calibrated at regular intervals (usually ranging from 1-12 months)
Testing facility shall have Standard Operating Procedures (SOPs) adequate to insure the quality & integrity of the data generated in the course of a study. All deviations from SOPs shall be authorized by the study director & documented in the raw data.
SOP for animal care: SOPs are required for all aspects of animal care. Newly received animals shall be isolated & health status evaluated. Animals shall be free of any disease or condition that might interfere at beginning of study. Animals of different species shall be housed in separate rooms. Feed & water are analyzed periodically for contaminants. Contaminant analysis of food & water for each & every study is not a requirement nor is analysis for a laundry list of contaminants.

This table summarized the GLP regulations and what tools are used to manage them.
GLP Regulations (Rules) | Documentation (Tools) |
Organization & Personnel | Training records, CVs, GLP Training |
Facilities | Maintain adequate space/ separation of chemicals from office areas |
Equipment | Calibration, logbooks of use, repair and maintenance; check freezers |
Facility Operation | Standard Operating Procedures |
Test, Control & Reference Substances | Chemical and sample inventory, track expiration dates, labeling |
Records and reports | Timely reporting, storage of raw data & reports |
Laboratory management responsibilities and organizational requirements take up about 15% of the GLP text, clearly demonstrating that the regulators also consider these points as important. Management has the overall responsibility for the implementation of GLP including both good science and good organization within their institution.
GLP Personnel should have sufficient education, training, and/or experience. They have access to protocols and SOPs. They are required to wear appropriate clothing. They avoid contamination through maintaining personal sanitation and health precautions. They are required to report if they are sick.
FDA regulations that require management responsibilities. The management needs to designate a Study Director and replace promptly if necessary. They need to assure there is a Quality Assurance Unit (QAU). They also need to assure that personnel, resources, facilities, equipment, materials, and methodologies are available as scheduled. They need to make sure that the personnel clearly understand the functions they are to perform and corrective actions are taken and documented as a result of deviations noted by the QAU. All the test and control article must be characterized.
The GLP regulation had a worldwide impact. Primary reason for that was world’s 30% of the pharmaceutical product market was and continues to be in USA.
OECD – Organization for Economic Cooperation and Development. The OECD is a worldwide organization dedicated to economic development.
JMAFF – Japanese Ministry of Agriculture, Forestry and Fisheries
Test site is Only defined by OECD and JMAFF. It is the location(s) at which a phase of the study is conducted.
Test Site Management is defined by OECD and JMAFF. It refers to the person(s) responsible for ensuring that the phase(s) of the study, for which he is responsible, are conducted according to these Principles of GLPs.
A term not defined by FDA or EPA, is Principal Investigator.
When the OECD regulations were updated in 1997, the term Principal Investigator was made a legal term. Only OECD and JMAFF regulations define the term Principal Investigator. An individual who, for a multi-site study, acts on behalf of the Study Director and has defined responsibility for delegated phases of the study
The Study Director’s responsibility for overall conduct of the study cannot be delegated to the Principal Investigator(s)
While the Principal Investigator is responsible for the portion of the study that is delegated to them, the Study Director is still the central point of control and must be notified of all study occurrences.
In the larger scheme of Drug Development, Good Laboratory Practices are the first set of regulations to be followed even before filing of an Investigational New Drug (IND) application, along with sometime Good Clinical Practice (GCP) and Good Manufacturing Practices (GMP). Once a candidate drug has passed through all clinical trials, the manufacturers can file New Drug Application for a drug to be marketed for medical purposes.
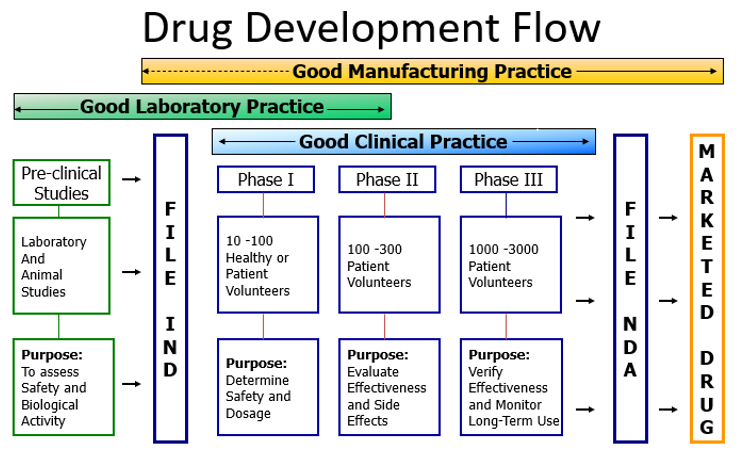
SUMMARY
Good Laboratory Practices apply to
- Non-clinical studies conducted
- It is for ensuring assessment of safety/ efficacy of chemicals/ pharmaceuticals.
- It is in compliance with regulatory agency guidelines
LINK
FDA CFR Title 21 Part 58- Good Laboratory Practices for Nonclinical Laboratory Studies
Standard Operating Procedures
Chapter 5: Standard Operating Procedures
Standard Operating Procedures (SOP) are a set of step-by-step instructions in a process or making a product to achieve a predictable, standardized, desired result.
SOPs are essentially backbone for achieving GLP, GMP and/or GCP using Quality Management System. Without Standard Operating Procedures, things can get complicated and confusing.

The need for SOPs: The FDA has placed us in an environment of regulatory compliance. Most regulatory and accrediting agencies require that those who perform procedures have the education, experience and training to do so.
SOP is mentioned in several guidelines as stated here.
Good Laboratory Practice 21 CFR 58.81(a)
A testing facility shall have standard operating procedures in writing setting forth nonclinical laboratory study methods that management is satisfied are adequate to ensure the quality and integrity of the data generated in the course of a study.
Good Manufacturing Practice 21 CFR 211.100
There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess.
Good Tissue Practice 21 CFR 1271.180
You must establish and maintain procedures appropriate to meet core CGTP requirements for all steps that you perform in the manufacture of HCT/Ps. You must design these procedures to prevent circumstances that increase the risk of the introduction, transmission, or spread of communicable diseases through the use of HCT/Ps.
ICH Guidance For Industry
E6 Good Clinical Practice: Consolidated Guidance
Principles of ICH GCP § 2.13
Systems with procedures that assure the quality of every aspect of the trial should be implemented.
SOPs can have different formats as different agencies, institutions, and companies may write them in different ways. The employees are trained on the SOP format and must enforce it.
A good SOP should always be:
- Accurate
- Up to Date
- Easy to understand and follow
- Accomplishes the purpose for which is written
SOPs are the foundation of training and have specific purpose:
- To provide people with all the information necessary to perform a job properly (i.e. a training tool)
- To ensure that the procedures are performed correctly and consistently
- To ensure compliance with university and government regulations
- To serve as a checklist for auditors
- To serve as an explanation of steps in a process so they can be reviewed in accident investigations.
- To serve as a historical record of the how, why and when of steps in an existing process occurred (for inspectors and attorneys)
- To Ensure Safety
- Maximize operational and production requirements
- To Ensure Consistent Training
- To Ensure Correct and Consistent Performance
- To Ensure Regulatory Compliance
- To Ensure Consistent Training
- To Ensure Correct and Consistent Performance
- To Ensure Regulatory Compliance
- Just Because It Makes Good Sense
Elements of SOP
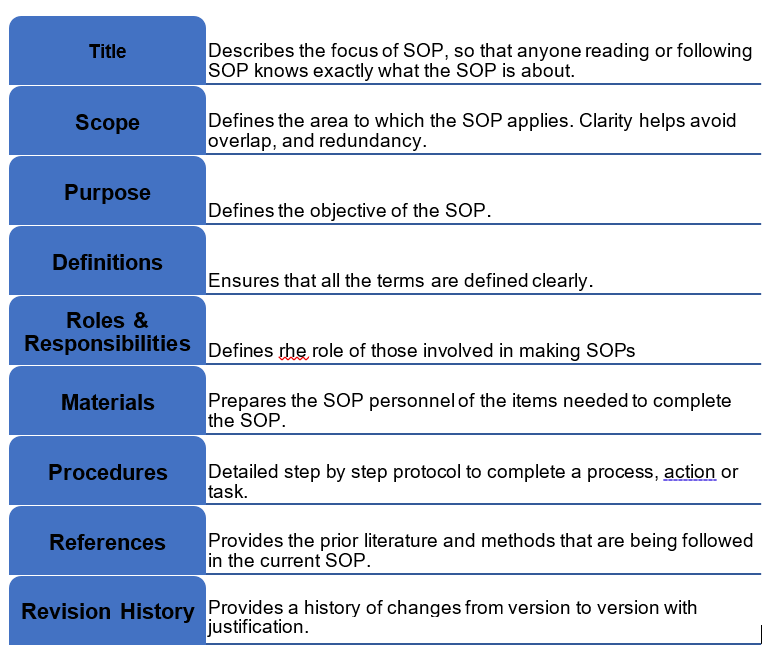
SOPs must always begin with active verbs like, begin, enter, start, analyze, submit, etc. Also if there are any forms, logs or other documents they should be attached to SOP. Some examples of the attachments are coversheet text for document approval, authorized copy log template, staff training documentation record or annual review coversheet.
Revision history of an SOP is extremely important. In includes what has changed and when. It is helpful specially in case of inspections, accidents or any legal implications.
Example of SOP Revision
SOP is an important element of FDA inspection.
|
Good Clinical Practices
Good Clinical Practice/ GCP is defined as an international standard for the design, conduct, performance, monitoring, auditing, recording, analysis and reporting of clinical trials or studies. GCP compliance provides public assurance that the rights, safety and well-being of human subjects involved in research are protected.
GCP is an international quality standard that is provided by the International Conference on Harmonisation (ICH). ICH-GCP harmonizes technical procedures and standards; improves quality; speeds time to market and decreases the cost to sponsors and the public.
Infamous cases such as Nazi physicians conducted large-scale trials on unwilling prisoners during World War II, black American men in syphilis studies (1932 –1972) who were not provided information on the study. This was followed by the declaration of Helsinki Agreement between countries that there needed to be a global standard by which all trial are conducted.
This is Good Clinical Practice – protects those in a trial, but also those who’s treatment will depend on the data. It essentially ensures that the rights of the patient are protected and by all those given a drug or intervention in the future based upon that data.
There are several acts/ laws that laid the foundation of GCP, The Nuremberg Code (1947), The Declaration of Helsinki (1964), The Belmont Report (1979), International Conference on Harmonisation (ICH-GCP), International Standards Organization 14155 and Code of Federal Regulations.
In 1997, the FDA endorsed the GCP Guidelines developed by ICH. ICH guidelines have been adopted into law in several countries (UK/ Europe), but used as guidance for the FDA in
the form of GCP.


Goals of GCP are to adhere to ethical standards while generating quality data by:
- Protecting the rights, safety and welfare of humans participating in research
- Assuring the quality, reliability and integrity of data collected
- Providing standards and guidelines for the conduct of clinical research
The steps to following a GCP in a protocol include:

Other things to think about while writing and SOP include clinical trial insurance / non-negligent harm cover, safety reporting, ethics committee safety and annual updates, clinical trial registries, sponsor reports, publication planning, logistics, transport, budgeting, drug/vaccine storage, sample transportation, export, storage, data archiving and maintaining SOP’s, training records and equipment service contracts.
ICH-GCP has 13 core standards:
Ethics 1. Ethical conduct of clinical trials 2. Benefits justify risks 3. Rights, safety, and well-being of subjects prevail Protocol and science: 4. Nonclinical and clinical information supports the trial 5. Compliance with a scientifically sound, detailed protocol Responsibilities: 6. IRB/IEC approval prior to initiation 7. Medical care/decisions by qualified physician 8. Each individual is qualified (education, training, experience) to perform his/her task Informed Consent: 9. Freely given from every subject prior to participation Data quality and integrity: 10. Accurate reporting, interpretation, and verification 11. Protects confidentiality of records Investigational Products 12. Conform to GMP’s and used per protocol (Quality Control/Quality Assurance) 13. Systems with procedures to ensure quality of every aspect of the trial. |
In order to ensure compliance with GCP, several partners are required:
- Regulatory Authorities who review submitted clinical data and conduct inspections
- The sponsor Company or institution/organization which takes responsibility for initiation, management, and financing of clinical trial
- The project monitor who is usually appointed by sponsor
- The investigator who is responsible for conduct of clinical trial at the trial site. Team leader.
- The pharmacist at the trial location is responsible for maintenance, storage and dispensing of investigational products eg. Drugs in clinical trials
- Patients are the human subjects
- Ethical review board or Committee for protection of subjects are appointed by Institution or if not available then the Authoritative Health Body in that Country will be responsible
- Committee to monitor large trials that overseas Sponsors eg. Drug Companies.
Any medical product/ device that comes to the market research studies have to be conducted to collect data on usual and unusual events, conditions, & population groups, to test hypotheses formulated from observations and/or intuition and to understand better – improve health outcomes with change. There are different types of medical research studies:
- Non-directed Data Capture
- Vital Statistics
- Directed Data Capture & Hypothesis Testing
- Cohort Studies, Case Control Studies
- Clinical Trials
- Investigation of Treatment/Condition
- Drug Trials
A properly planned and executed clinical trial is a powerful experimental technique for assessing the effectiveness of an intervention. Clinical trial is different from ‘Standard of Care’
- Involves human subjects
- Test an ‘intervention’ – be it a product, procedure or health care sytem….in order to improve standard of care
- Measures effects over a period
- Most have a comparison CONTROL group
- Must have method to measure intervention
- Focuses on unknowns: effect of intervention
- Must be done before medication is part of standard of care
- Standard of Care all about clinical judgement decision/flexibility – trials need all to stick with the protocol, no deviation – within your clinical judgement
Examples of clinical trials
They could be small investigator-led fellowship type studies that are addressing a disease management question, through to large multi-center programs within collaborations or with product development sponsors assessing new products for licensure. Clinical trials may be dealing with Improving disease management in very sick children such as severe malaria, malnutrition and management of seizures and in-patient trials for product development such as PK studies. It could be phase II and III regulatory trials in drug and vaccines for malaria and HIV.
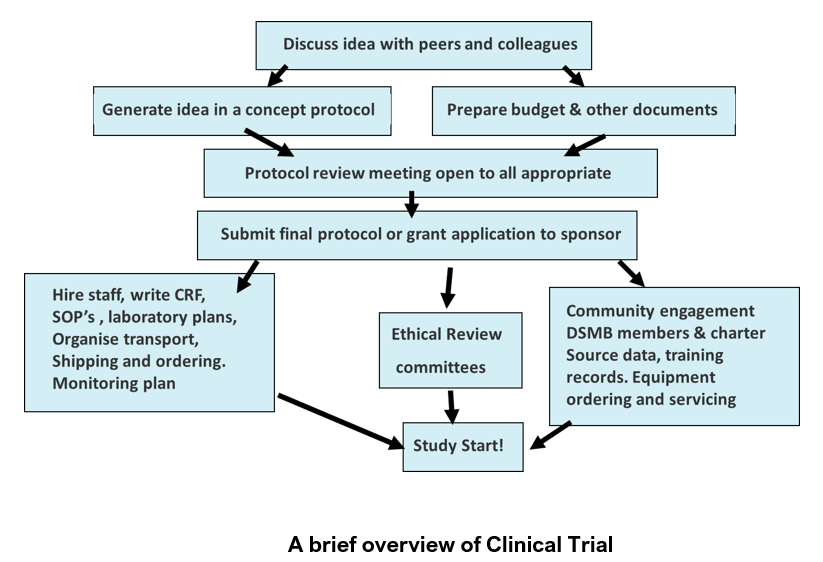
SOPs in Clinical Research
International Conference on Harmonization (ICH) defines a SOP as “Detailed, written instructions to achieve uniformity of the performance of a specific function.” (ICH GCP 1.55).
In simple terms a SOP is a written process and a way for the clinical site to perform a task the same way each time it is completed.
SOPs are not specifically mentioned in the FDA regulations. However, there is guidance and regulations that infer responsibility and SOPs formalize investigator responsibilities.
21 CFR312.53 of FDA states that the investigator will “ensure that all associates, colleagues, and employees assisting in the conduct of the study(ies) are informed of their obligations in meeting the above commitments.”
Some of the examples of SOP topics include, preparing and Submitting Initial IRB (Institutional Review Board) documents, preparing and submitting continuing review IRB documents, preparing and submitting amendment IRB documents, establishing and training the clinical study team, and delegating responsibilities, establishing study files and establishing Source Documents
Benefits of SOP in clinical trials include:
- Ensures that all research conducted within the clinical site follows federal regulations, ICH GCP, and institutional policies to protect the rights and welfare of human study participants.
- Provides autonomy within the clinical site.
- Improves the quality of the data collected, thereby improving the science of the study.
- Utilized as a reference and guideline as to how research will be conducted within the clinical site
- Excellent training source for new employees and/or fellows
Elements of SOP
Just like SOPs discussed in chapter 5, the potential elements of the SOP related to clinical trials include
- Header – title, original version date, revision date, effective date, approved by
- Purpose – why one has the policy
- Responsibilities – who the policy pertains to
- Instruction/Procedures – how to accomplish the items of the policy
- References – what the policy is based on
- Appendix – source documents/case report forms
SOPs should be written in clear, concise language, use active voice, avoid names and use titles instead. The process mapping for writing SOPs include determining which clinical site task needs mapping, laying out all the steps currently used to complete that task. “Mapping” involves taking each step in the task and making it more efficient and easier to follow.
Once the process of mapping is finished, the process map is converted to an outline for easy use. Once a task has been mapped, it should be tested.
Also, the implementation and monitoring of SOPs should be introduced gradually, prioritizing the most relevant SOPs and present them first. Principle Investigator should approve all SOPs and designate an effective date. SOPs should be reviewed on a regular basis (usually annually) to ensure policy based regulations are up-to-date. Previous versions of SOPs should be retained. All staff should have SOP training. Training should be documented. SOP should be accessible to staff.
Process Mapping for Making a Cup of Coffee
The process mapping for making a cup of coffee can be divided in primary and secondary steps. One advantage of a two-tiered system is that SOPs will rarely need to be changed, whereas guidelines may need to be changed or updated more frequently due to changes in organizational structure or equipment.
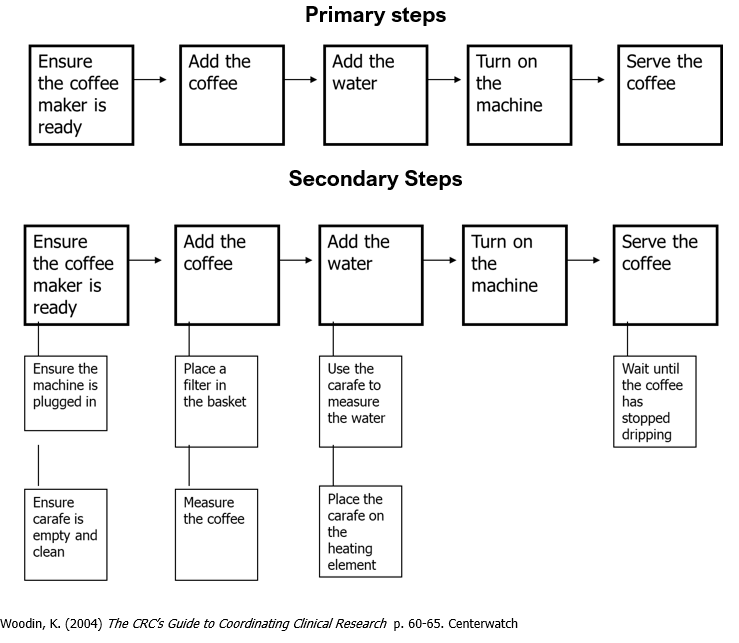

1.0 Purpose
To ensure company employees wanting coffee do so appropriately and according to clinical site standards.
2.0 Responsibilities
Clinical site personnel who want to make coffee.
3.0 Instructions
3.1 Ensure coffee maker is plugged in and the carafe is clean and empty.
3.2 Place a filter in the coffee receptacle and add the appropriate amount of coffee.
3.3 Fill the carafe with the desired level of water and pour into the water reservoir.
3.4 Place the carafe on the heating element and turn the machine on.
3.5 When the coffee has stopped dripping into the carafe, it is ready to serve.
SOP vs MOP
Standard Operating Procedure (SOP) and a Manual of Procedures (MOP) terms are used interchangeably.
SOP provides general information that is to be utilized throughout any research study.
MOP is specifically written for a particular research study which will incorporate elements of the SOP.
- The MOP should be written so that anyone in your clinical site can follow the procedures for that study and find all relevant materials.
- The MOP should be extremely detailed.
Both documents are important. It starts with the SOP first. Once the general procedures are written, the MOP will be easier. This process takes a lot of time and patience. Both SOP and MOP provide standardization processes.
Good Manufacturing Practices
Good Manufacturing Practice (GMP) is a set of regulations, codes, and guidelines for the manufacture of drug substances and drug products, medical devices, in vivo and in vitro diagnostic products, and foods.
GMP practices are enforced worldwide. Good Manufacturing Practices are enforced in the United States by the FDA. In the United Kingdom they are enforced by the Medicines and Healthcare Products Regulatory Agency, in Australia by the Therapeutically Goods Administration, in India by the Ministry of Health, multinational and/or foreign enterprises. Many underdeveloped countries lack GMPs.

History of GMP
The beginning of modern standards of GMP can be traced to an incident in 1941, when the Winthrop Chemical company of New York put Sulfathiaziole tablets in market that were contaminated with phenobarbital. This resulted in hundreds of death. There was a huge challenge in investigation into production as well as pulling the remaining drug off the market. This led FDA to enforce and revise manufacturing and quality control requirements and GMP was born in 1941 (PDA J Pharm Sci Technol .1999;53(3):148-53)
Then in 1962, GMP was strengthened by Kefauver-Harris Drug Amendments, after Thalidomide tragedy. Thousands of children born with birth defects due to adverse drug reactions of morning sickness pill taken by mothers. FDA introduced “Proof of efficacy” law. This strengthened FDA’s regulations regarding experimentation on humans and proposed new way how drugs are approved and regulated.
In 1972 and 1973, pacemaker failures were reported. In 1975 - hearing-Dalkon Shield intrauterine device caused thousands of injuries. In 1976, medical device amendments was introduced. According to that medical devices were classified as Class I, II and III based on degree of control necessary to be safe and effective.
In 1978 , one of the major manufacturer of infant formula reformulated two of its soy products, this resulted in infants being diagnosed with hypochloremic metabolic alkalosis. The Infant Formula Act was passed in 1980 by the Congress. It established nutrient and quality control procedures, prescribes recall procedures and specified inspection requirements. This the act ensured greater regulatory control over the formulation and production of infant formula.
GMP
Good manufacturing practices (GMP) are the practices required in order to conform to the guidelines recommended by agencies that control authorization and licensing for manufacture and sale of food, drug products, and active pharmaceutical products.
GMPs are enforced in the United States by the U.S. Food and Drug Administration (FDA), under Title 21 CFR. FDA ensures good manufacturing practices through various guidance in code of federal regulations (CFR).
Good manufacturing Practices were implemented by the FDA to ensure that Drug Products are manufactured in an appropriate and safe way.
(21 CFR 210 & 211)
FDCA grants FDA authority to ensure compliance with current GMP
(cGMP) (§ § 301(a), 501(a)(2)(b); 21 USC § § 331(a); 351(a)(2)(b)
A Drug is adulterated if found not to be manufactured according to cGMP’s
(§ § 301(a), 501(a)(2)(b)
A drug can meet its specifications, contain no detectable impurities, be both safe and effective and still be considered adulterated if the manufacturing was not in compliance with cGMP’s.
(FDCA §501 (a)(2), 21 U.S.C. §351 (a)(2)(b), 21 CFR Part 210 & 211);
Quality should be built into the product and testing alone cannot be relied upon to ensure product quality”
(FDA Guidance for Industry, Sept. 2006)
The role of the FDA is to determine which practices in use are “good” and enforce those. FDA does not prescribe “how to make products”, but requires manufacturers to do so. Companies must establish procedures which assure the drug is manufactured in a way to consistently meet specifications.
cGMP
GMP is always written as current Good Manufacturing Practices (cGMP). Good manufacturing practices are required to conform to the guidelines recommended by agencies that control the authorization and licensing of the manufacture and sale of food and beverages, cosmetics, pharmaceutical products, dietary supplements, and medical devices. The word current or “c” reminds the manufacturers that they must employ technologies and systems that are up-to-date in order to comply with the regulations. (https://ispe.org/initiatives/regulatory-resources/gmp/what-is-gmp)
FDA and other regulatory agencies are authorized to conduct inspections that can be scheduled in advance or unannounced. When a company adheres to cGMP regulations, consumers can be assured that the identity, strength and purity of drug products have been tested to a high standard.
If a drug made by a certain company falls short of standards, FDA will advise company to recall the product. If a company refuses to recall a drug, FDA can issue warning to the public, seize the product and bring the company to court which could result in fines and jail time.


Regulation of Drug Manufacturing
The worldwide pharmaceutical market is projected to be around $1.5 trillion in 2021 and growing at the rate of nearly 6.4 % (https://markets.businessinsider.com/news/stocks/the-mid-size-pharmaceutical-market-market-assessment-growth-forecasts-and-strategic-consolidations-for-key-players-1027168940) . The pharmaceutical industry can be divided into three sectors based on the revenue – large, mid-size and small pharma companies. Small and mid-size companies could be privately or publicly held and make everything from topical to high-tech bipharmas and orphan drugs. Some of the challenges that small and mid-size drug companies may have is in (1) Understanding drug current Good Manufacturing Practices (cGMPs), and defining "adulteration" and "misbranding" (2) Review the different types of inspections (3) Explore the elements of a 483 observation and the components involved with closing out an inspection.
Two major challenges that the companies can have are:
Adulteration---methods or processes used are not in conformance with good manufacturing practices (FDCA §501 (a)(2), 21 U.S.C. §351 (a)(2)(b), 21 CFR Part 210 & 211);
Misbranding--- provisions can pertain to materials that are physically distant from the product or its container (ads making improper claims run in journals). Labeling is false or misleading (FDCA §502 a), 21 U.S.C. §352(a));
The companies require written procedures (SOP’s and validation) to conduct batch production and processing in a way ensuring the accuracy of: (1) Identity (2) Strength (3) Purity (4) Quality (21 CFR 211). They also need attorneys to review to ensure compliance with latest Policy/Regulations of FDA.
Validation Process
The foundation for process validation is provided in § 211.100(a), which states that there shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess.
- The cGMP regulations require that manufacturing processes be designed and controlled to assure that in-process materials and the finished product meet predetermined quality requirements and do so consistently and reliably.
- The regulation requires manufacturers to design a process, including operations and controls, which results in a product meeting these attributes.
- The goal of validation is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture.
- A system or systems for detecting unplanned departures from the process as designed is essential to accomplish this goal.
FDA Inspection
FDA may conduct inspection of a facility for a scheduled investigation, survey or in response to a reported problem. This is to assess whether the regulated facilities are complying to the laws and regulation as per Food, Drug and Cosmetics Act and related Acts.
During inspection, FDA investigator presents their identity and a “Notice of Inspection” which is also called as FDA form 482. A designated company personnel typically accompanies the investigator during the inspection. The investigator may examine the production process of a company and may call check records and collect samples. At the end of the inspection of the manufacturing facility, FDA investigator will discuss his findings and also leave list of “Inspectional Observations” in FDA form 483. An FDA Form 483 is issued to firm management at the conclusion of an inspection when an investigator(s) has observed any conditions that in their judgement may constitute violations of the Food Drug and Cosmetic (FD&C) Act and related Acts.
This facility then has to respond to the FDA-483 stating appropriate responses and corrective action as needed. FDA expects a response from manufacturer on each observation in15 business days. The company must explain the problem identified, propose action or plan to prevent reoccurrence of observation and use system preventative answers.
FDA is entitled to relevant information concerning whether drugs (for human consumption) are manufactured, transported, processed or packed (inn accordance with the Act [FDCA] (704(a)(1); 21 USC 374(a)(1))
FDA is not entitled to (1) Financial Data (2) Sales Data (other than shipping amounts) (3) Pricing data (4) Personnel data (other than the qualifications of professional personnel) (5) Research data (except for data required to be disclosed under 21 USC 374 ‘approval data’) (6) Internal audit (7) supplier audit and (8) management reviews.
FDCA does not authorize investigators to interview a company’s employees; (if so, with counsel present)
cGMP violation includes finding of adulteration based on failure to comply with GMP’s even if product quality is fine (Title 21 S. 351(a)(2)(B)). Even if the final product is “pharmaceutically perfect,” manufacturing non-compliance will deem product adulterated. GMP has to to be not only “Good” but “Current”. Hence the importance of cGMP in the manufacturing industry.
Introduction to biohazard – Risk Management
Working safely while performing experiments involving biohazards requires merging the safety practices required when working with hazardous chemicals with some new practices that are tailored to the special risks associated with biological hazards. Safe laboratory practice requires knowledge of the risks associated with hazards and a plan to manage those risks.
Biohazard
A biohazard is a biological agent with the potential to cause harm in humans. There are numerous different classes of biohazards used in the biotechnology laboratory. Such biohazards include bacteria, viruses, fungi and even smaller particles such as prions, which are biohazardous forms of naturally occurring proteins. Within each of these classes are varying degrees of risk, and it is your job to learn how to manage those risks to protect your safety and that of your laboratory co-workers.
Pathogenicity
Pathogenicity is the ability of an organism to cause disease in humans or other host organisms. Pathogens include bacteria, fungi, viruses, and other biological entities. The practice of safe handling of pathogens and their toxins in the laboratory is accomplished through the application of containment principles and risk assessment. Containment means the use of laboratory practice and technique, safety equipment, and facility design to reduce the exposure of laboratory workers to pathogens.
Carriers
If an agent infects a host but does not cause disease in that host, the host is a carrier. A carrier is an infected individual capable of spreading infectious agents to others. They are termed as carriers because they are asymptomatic, often play important roles in the spread of disease through a population.
Super-spreaders are people who seem to transmit a given infectious disease significantly more widely than most.
Routes of Infection
Pathogens are, by nature, infectious, meaning that they are able to invade a host. This invasion can occur through a number of routes such as (1) inhalation (2) skin/ eye contact (3) ingestion (4) injection.
The infectious agents may enter the body through the respiratory system, via the process of inhalation, as is common with cold and flu viruses. An example of this is the spread of COVID19 by SARS-CoV2 virus.
Second, the agent may be introduced via the skin through contact with eyes and other mucous membranes, as is seen when bacterial conjuctivitis is spread.
Third, the agent may enter through the mouth and the digestive system, as occurs with food-borne illnesses such as salmonella, and water-borne.
Finally, the agent may penetrate the body through injection. Common forms of injection include insect bites and stings.
Biohazardous Agents
The same routes of entry are employed by biohazardous agents used in the laboratory.
- In the laboratory, the most likely route of infection is through the inhalation of bioaerosols, which are sprays of small infectious particles suspended in air.
- In fact, 70% of all laboratory-acquired infections are the result of bioaerosols.
- Many of the safety guidelines mentioned for chemical handling apply to biohazard work. But there are some special considerations when working with biohazards.
- Imagine that you accept a position in a company that is working on a vaccine for the virus that causes Hepatitis C, one of the most common blood-borne agents in the US. You recognize that this is a biohazard, but there are a number of questions that you should ask prior to beginning work with this or any biohazard.
- First, ask whether this is a know human/ primate pathogen. If you know that a particular biohazard has been shown to cause biological harm in humans or other primates, then you know that you will need to take particular care. Of course, you will then need to know what risks are associated with this hazard.

Special considerations when working with biohazards are
- Pathogenic
- Associated risks
- Laboratory-acquired infections
- Treatments
- Allergies
Imagine that you accept a position in a company that is working on a vaccine for the virus that causes Hepatitis C, one of the most common blood-borne agents in the US. You recognize that this is a biohazard, but there are a number of questions that you should ask prior to beginning work with this or any biohazard.
First, ask whether this is a known human/ primate pathogen. If you know that a particular biohazard has been shown to cause biological harm in humans or other primates, then you know that you will need to take particular care. Of course, you will then need to know what risks are associated with this hazard. For instance, Hepatitis C can cause cirrhosis of the liver. Since your liver carries out essential functions, then you will know that infection with this virus could prove fatal. Other biohazards may have considerably less serious risks associated with infection. Once you have determined what the potential outcomes of infection may be, you should consider whether this biohazard been associated with lab-acquired infections, and if so, with what health consequences. Considering that infection with the biohazard is a possibility, you should investigate whether there is either a treatment for the disease, or if, in fact, a vaccine is available that would prevent you from becoming infected in the first place. Of course, if there is a vaccine, that does not mean that the organism is not longer a serious hazard. Remember that vaccines are not effective all of the time! For instance, the virus that causes rabies can be prevented via vaccination; however, since rabies is a nearly invariably fatal illness if contracted, you would not want to handle the rabies virus in a careless way, even though you would, presumably have been vaccinated. Next, question whether any allergies are induced by this agent. For instance, various molds are known to be very allergenic.
Biohazard considerations may include (1) Infectiousness of biohazards may vary greatly. (For instance, were you to accidentally stick yourself with a needle contaminated with Hepatitis B virus, you would be ten times as likely to become infected than if the needle were contaminated with HIV.) (2) Exposure may be limiting or more. (3) Special safety precautions (Will work occur in a biological safety cabinet, can you avoid using needles, and thereby reduce the likelihood of injection of the biohazard?) (4) Is the level of risk of working with this agent acceptable to the worker? (5) Do you need special training before work begins?
Laboratory-acquired Infections (LAI)
Laboratory-acquired infections can be traced directly to lab organisms handled by or used in the vicinity of the infected individuals. Thousands of laboratory acquired infections have been documented, resulting in hundreds of deaths. 1/4 of these cases were found in non-researchers such as dishwashers, custodians, office staff. Your actions in the laboratory affect your co-workers. If biohazards are not managed correctly, then the biohazard can be spread throughout the laboratory and then, eventually, even outside of the laboratory. The exposure of the public to biohazardous agents must absolutely by avoided.
Standard Microbiological Practices
Practices to be used when working with all microbiological organisms are meant for protecting workers. It is also to protect culture from contamination.
Standard Microbiological Practices are designed to protect you from your laboratory culture and also to protect your laboratory culture from you!

These rules are designed to prevent the biological organism from gaining access to your body through any of the the four routes available: inhalation, skin/eye contact, ingestion or injection. Simple rules of good laboratory hygiene will go a long way towards preventing any contamination. If any contamination has occurred, it is critical to decontaminate workspaces after each laboratory session, and to always wash hands thoroughly with soap and water prior to leaving the lab!.
Biocontainment is the control of biohazards through:
- Practices & procedures, including administrative controls
• Good lab practices
• Written SOPs for research activities, specialized equipment, etc
• Required training
- Primary barriers – Physical barriers or personal protective equipment between lab worker and pathogen
• Biosafety cabinets (BSCs)
• Lab equipment (pipetting devices, waste containers, safety centrifuge cups)
• Personal protective equipment
- Secondary barriers are structural aspects of the laboratory that make working environment safer against infection.
• Building & room construction – the floor plan
• HVAC issues – directional airflow, filtration, waste treatment
Universal Precautions
Universal precautions developed to protect health professionals. They most often apply in a clinical setting. They are also important for field epidemiology practices during an outbreak investigation (e.g., collecting lab specimens)
These precautions include hand hygiene, gloves, gown, masks, eye protection, face shields, safe injection practices. They require that all equipment or contaminated items are handled to prevent transmission of infectious agents.
Biosafety Guidelines
While we consider safe handling of biohazards, governmental organizations sit at the top of the hierarchy and create rules and standards for working safely with biohazards. The two most prominent governmental agencies involved are the Center for Disease Control and Prevention (CDC) and the National Institute of Health (NIH). Other organizations also publish standards for workers to use to reduce risk associated with the use of biohazards in the laboratory. |
Biosafety Concepts
Biosafety in Microbiological and Biomedical Laboratories (BMBL) is an advisory document published by NIH and CDC. It recommends best practices for the safe conduct of work in biomedical and clinical laboratories. BMBL hasbecome the cornerstone of the biosafety practices in US and many countries around the world. (https://www.cdc.gov/labs/pdf/CDC-BiosafetyMicrobiologicalBiomedicalLaboratories-2009-P.PDF). |

Importance of Biosafety
Laboratorians have long recognized the hazards of processing infectious agents and the importance of biosafety. In response to these hazards, guidelines have been developed to protect workers in microbiological and medical labs through a combination of safeguards including engineering controls, management policies and work practices.
In any laboratory, precautions must be taken so that the people researching or trying to identify organisms do not become infected themselves. According to the Centers for Disease Control and Prevention (CDC), scientists and lab technicians have to be very aware of microorganisms; while handling or testing clinical specimens, they could accidentally infect themselves or their coworkers. Because of this danger, labs must adhere to very specific safety regulations to work with organisms that pose a threat to human health. The biosafety levels are hence precautionary measures so people researching or trying to identify organisms do not become infected.
Biosafety Level Sign
BMBL requires that a biohazard sign be posted at the entry of laboratory when the biohazardous material (infectious agents) are present in the lab. Example of biosafety sign posted outside lab working with infectious agent.

Biosafety Levels
A biosafety level describes the microbiological techniques, lab practices, safety equipment and lab facilities necessary to protect workers and the environment from biohazardous material.
There are four basic biosafety levels as determined by CDC and NIH. Regulations outline precautions, special practices, decontamination procedures.
BSL-1 – Minimal potential hazard to lab personal & environment.
BSL-2 – Moderate potential hazard.
BSL-3 - Serious potential hazard.
BSL-4 – Life threatening hazard.
The regulations outline precautions, special practices, and decontamination procedures for labs that work with infectious agents. Based on the degree of hazard posed by these agents, labs are divided into four biosafety levels, and mandated protective practices increase with each level. Biosafety Level 1 labs work with the least dangerous agents and require the fewest precautions; Biosafety Level 4 labs have the strictest methods for handling organisms because they deal with agents that are most dangerous to human health.

Risk Groups
In many countries, including the United States, biological agents are categorized in Risk Groups (RG) based on their relative risk. Depending on the country or organization, this classification system might take the following factors into consideration:
- Pathogenicity of the organism
- Mode of transmission and host range
- Availability of effective preventive measures (e.g., vaccines)
- Availability of effective treatment (e.g., antibiotics)
- Other factors
It is important to understand that biological agents are classified in a graded fashion such that the level of hazard associated with RG1 being the lowest and RG4 being the highest. EHS Biosafety follows the NIH Guidelines categorization of Risk Groups as follows:
- RG1 – Are not associated with disease in healthy adult humans or animals
- RG2 – Are associated with disease which is rarely serious and for which preventative or therapeutics is often available
- RG3 – Are associated with serious or lethal human disease for which preventative or therapeutics may be available
- RG4 – Are associated with lethal human disease for which preventative or therapeutics are not readily available
Risk Groups and Biosafety Levels
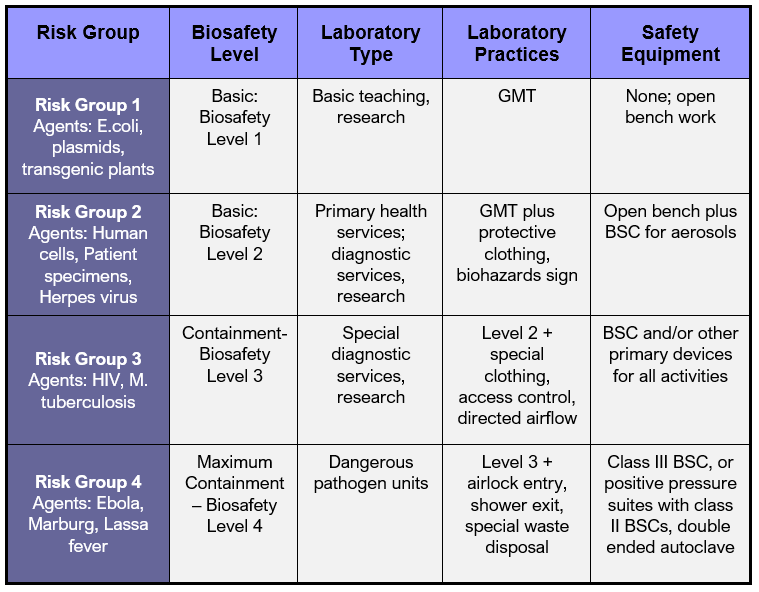
Biosafety Level 1 (BSL-1)
Biosafety Level 1 agents pose no threat to human health; that is, they are not known to cause disease in healthy adults. Some of these organisms may be known to cause disease in immunocompromised individuals.
This is the type of laboratory found in municipal water-testing laboratories, in high schools, and in some community colleges.
Agents studied in BSL-1 labs include Bacillus subtilis, Naegleria gruberi, infectious canine hepatitis virus, and non-pathogenic E. coli species (see Figure 1). (2)

Important features of BSL-1 include
- Suitable for work involving well-characterized agents not known to consistently cause disease in immunocompetent adult humans.
- Minimal potential hazard to laboratory personnel and the environment.
- Laboratories are not necessarily separated from the general traffic patterns in the building.
- Work is typically conducted on open bench tops using standard microbiological practices.
- Special containment equipment or facility design is not required.
- Laboratory personnel must have specific training in the procedures conducted in the laboratory and must be supervised.
Biosafety Level 2 (BSL-2)
Agents associated with human disease are studied in BSL-2 laboratories. A BSL-2 lab is generally required for working with any human-derived blood, other bodily fluids (particularly when visibly contaminated with blood), or tissues in which the presence of an infectious agent may be unknown.
BSL-2 labs work with organisms such as the measles virus, many Salmonella species, pathogenic Toxoplasma species, Clostridium botulinum, hepatitis B virus (see Figure 2), and other bloodborne pathogens.
Examples of BSL-2 facilities may include local health departments, universities, state laboratories, private laboratories (hospitals, health care systems), industrial laboratories.

Important features of BSL-2 include:
- Builds upon BSL-1
- BSL-2 is suitable for work involving agents that pose moderate hazards to personnel and the environment.
- Laboratory personnel have specific training in handling pathogenic agents
- Personnel are supervised by scientists competent in handling infectious agents and associated procedures
- Access to the laboratory is restricted when work is being conducted
- All procedures in which infectious aerosols or splashes may be created are conducted in biological safety cabinets (BSCs) or other physical containment equipment.
Biosafety Level 3 (BSL-3)
The primary hazard for personnel working with BSL-3 agents is risk of infection from needle sticks, ingestion, or exposure to infectious aerosols. Some examples of the microbes that need to be handled in a BSL-3 lab are Mycobacterium tuberculosis, H1N1 flu, SARS virus, Rabies virus, West Nile virus.
For example, part of public health surveillance for West Nile virus (WNV), is testing birds for the presence of the virus since birds often serve as the first indicator of the virus in a geographic region or in a season. In August of 2002, a state laboratory worker accidentally cut his finger while dissecting a bird to test for WNV. Four days later, this worker had symptoms of fever, myalgia, recurring sweats, and hot flashes. The worker and the bird he was working with were both eventually diagnosed with WNV. (3) There were 2 lab-acquired cases of WNV in 2002.
Primary hazards include needle sticks, ingestion, exposure to infectious aerosols.
The risk posed to researchers by this highly infectious agent – severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) – currently dictates that live-virus research is performed under BSL3 conditions.
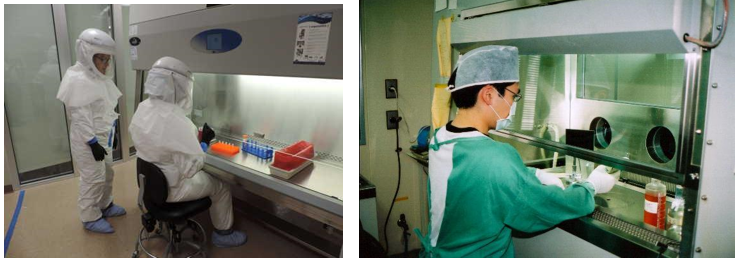
Important features of Biosafety Level-3 include:
- Is applicable to clinical, diagnostic, teaching, research, or production facilities where work is performed with indigenous or exotic agents that may cause serious or potentially lethal disease through inhalation route exposure.
- Laboratory personnel must receive specific training in handling pathogenic and potentially lethal agents
- Must be supervised by scientists competent in handling infectious agents and associated procedures.
- Biosafety Level 2 plus all procedures involving the manipulation of infectious materials must be conducted within BSCs, or other physical containment devices
- Personnel wear additional appropriate personal protective equipment including respiratory protection as determined by risk assessment
- A BSL-3 laboratory has special engineering and design features include directional air flow.
Biosafety Level 4 (BSL-4)
BSL-4 includes the organisms that pose the highest risk level to both the laboratory worker and the general public. BSL4 organisms are handled in glove boxes, or in biological safety cabinets by workers clothed in full body, air-supplied suits. BSL4 facilities are isolated from other facilities and must have their own dedicated air supply and exhaust. Workers change and decontaminate clothing upon exit. Rigorous training is required of those workers who operate in BSL4 facilities. As of 2010, only 13 operating or planned BSL4 facilities existed in the United States.
BSL4 organisms include the Ebola, Lassa and Marburg viruses.
BSL4 includes:
- Maximum containment facilities
- Pressurized Containment Suite
- BSL-3 + Class III Biosafety Cabinet
- Chemical decontamination showers
- Liquid effluent collection / decontamination
- Clothing change, and shower upon exit
Important features of BSL-4 include:
- Laboratory staff must have specific and thorough training in handling extremely hazardous infectious agents.
- Laboratory staff must understand the primary and secondary containment functions of standard and special practices, containment equipment, and laboratory design characteristics.
- All laboratory staff and supervisors must be competent in handling agents and procedures requiring BSL-4 containment.
- Access to the laboratory is controlled by the laboratory supervisor in accordance with institutional policies
- Two types of laboratory providing absolute separation of the worker from the infectious agents
- Suit Laboratory
- Cabinet Laboratory
Biological Safety Cabinets
When working with hazardous chemicals, especially those that are volatile, it is often necessary to use a fume hood. Likewise, technicians working with biohazards will often need to work in a type of hood called a biological safety cabinet. We will discuss biological safety cabinets briefly here. It is important to remember two things about biological safety cabinets. First, there are multiple types of biological safety cabinets, and you will need to know the specific type required by the biohazard with which you are working. Second, biological safety cabinets are not fume hoods and are not designed to remove chemical vapors.
In fact, biological safety cabinets are a form of physical containment designed to separate the worker from the biohazard. The organism is exposed to a flow of sterile air, which protects the organism from the worker and environment. The air flow is into the hood, and exhausted air is sterile-filtered to protect the worker and the environment from the biohazard. Biological safety cabinets can filter most particles, including most viruses, from the air.
Biosafety cabinets provides containment for aerosols and separates work area from operator and lab while providing clean air
- Able to filter most particles (including most viruses) from air
- Does not remove chemical vapors

Classes of BSC
Class I and Class II cabinets draw air from the room into the hood and look like fume hoods. Class III cabinets are glove boxes, which provide total containment. These glove boxes are fitted with air-tight gloves and have attached autoclaves, incubators and air locks. Many biological safety cabinets are also fitted with germicidal UV lamps which are turned on after work is complete and workers have left the room. These UV lamps decontaminate all work surfaces.
Regardless of the work that you are doing with biohazards, be sure that you are aware of the risks associated with the organism that you are using and that you receive appropriate training to work safely in the lab.

Animal Biosafety Levels 1-4 (ABSL)
Laboratory animal facilities
Animal models that support research
Guidelines for working safely in animal research facilities

Accidental Spills
- Evacuate area, alert personnel and cordon off so that aerosols may settle
- Don PPE; Cover with paper towels and apply bleach (1 part bleach: 9 parts water
- Allow 15 – 20 min contact time
- Wipe up working towards center
- Use tongs if broken glass is involved
- Is Recombinant DNA involved?

First Aid measures
- Every lab has a first aid box
- Splash to Eye or Needlestick Injury
- Rinse thoroughly for 15 minutes at the eyewash or sink.
Cleanroom Basics and Waste Management
A cleanroom is a controlled environment where pollutants like dust, airborne microbes, and aerosol particles are filtered out in order to provide the cleanest area possible. Most cleanrooms are used for manufacturing products such as biotech or pharmaceutical products, and medical equipment. A clean environment is designed to reduce the contamination of processes and materials. This is accomplished by removing or reducing contamination sources.
The purpose of clean room is to
- Promote Successful Cleanroom Operations
- Ensure Safety in the Clean Environment
- Provide Operational Conditions that Meet Process & User Needs
Successful cleanroom operation relies on each user’s understanding, participation and self discipline. The protocol provides basic awareness and general guidelines for cleanroom users. The success of each user relies on trust, understanding and shared responsibility among all users.
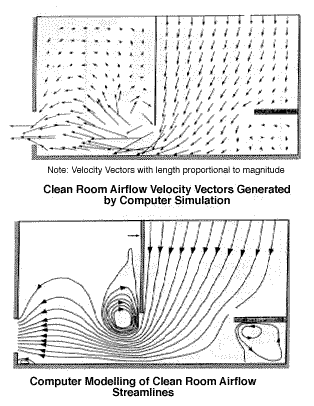
Principles of the clean environment
Types of contamination in a cleanroom include:
Major contamination sources are human (~75%), ventilation (~15%), room structure (~5%) and equipment (~5%). Humans are the biggest source of contamination. They generate >1x105 particles per minute when motionless (fully gowned). Environmental Control can be done by entrance and exit, materials and supplies, cleaning and maintenance and controlling atmospheric factors. Personnel Control can be done by dress code, personal hygiene and gowning. Dress code typically include no sleeveless shirts, no shorts or skirts, no slippers or sandals and no jewelry that can puncture garments or gloves. Personnel are required to avoid clothing that sheds. Personnel hygiene includes showering each day before entry, controlling dermatitis & dandruff, no smoking before entry, no chewing gum or tobacco, no Cosmetics should be worn and facial hair needs to be covered. Proper gowning order
Garments should not be removed from cleanroom unless in approved container Personnel cannot walk out of the cleanroom with your garments on. They are required to change garments when soiled or showing any visible signs of wear. They can’t reach inside your garment while in the cleanroom |
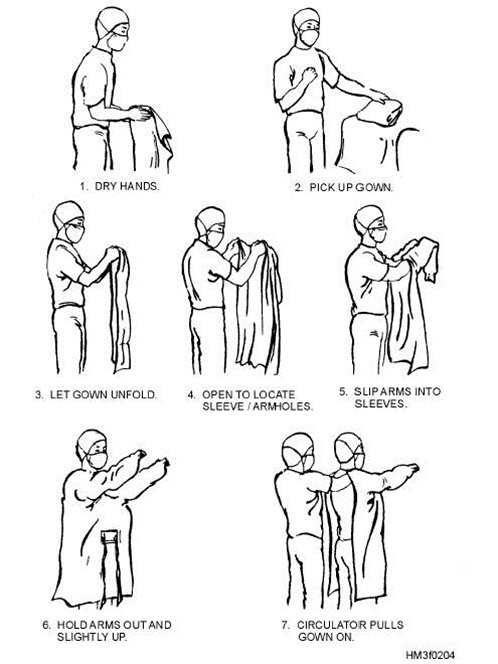
Garment use and storage
The garment that a personnel may wear can be reusable or non-reusable depending on the manufacturing facility and the products that are being manufactured.
Reusable garments should be stored in gown bin or zip lock bag. These may include coveralls, hood, knee-high Booties or safety glasses.
Non-reusable garments are thrown in trash. These include hair nets & beard covers, blue disposable shoe covers, gloves, face masks.
Personnel entry and exit: In a cleanroom, entry and exit should be done quickly. Only one person may enter at a time and use their own access card to do so always. They must pass from the gowning area to the clean area slowly to reduce migration of particles between areas.
Materials and supplies: As far as the materials and supplies transportation in and out of cleanroom, non-cleanroom items cannot be taken into the cleanroom and cleanroom items cannot be taken out of the cleanroom. Pencils or erasers cannot be used in the cleanroom. Paper should be kept in a plastic sleeve. Everything that goes into the cleanroom must be cleaned.
Chemicals: New chemicals cannot be taken into the cleanroom without permission. Personnel using any chemical in the cleanroom should be very familiar with the MSDS. Large quantities of chemicals must be stored outside the cleanroom. Chemicals inside the cleanroom should be properly stored. All chemical containers should be clearly labeled with their contents and Hazard Classification. Unattended chemicals and experiments should be labeled with the owner’s name, immediate contact number, list of all chemicals involved, and estimated time of return or completion.
Chemical Handling: In a cleanroom, safety carriers must be used with glass bottles 2L and larger. Chemicals should not be transported in open containers. Chemicals should be used inside approved hoods or under properly positioned snorkel extractors. Full apron, trionic gloves and full face shield are required for using all acids. Gloves, lab coat, and safety glasses are considered minimum personal protective equipment when handling any chemical.
In case of a small spill, all users must be informed in the cleanroom, the affected area should be clearly marked, appropriate absorbent material should be used to clean up the spill.
In case of large spill similar protocols are in effect. All users in the cleanroom must be informed of the spill. The affected area should be marked and all personnel should be evacuated. Environmental Health and safety offices must be informed immediately.
Handling of chemical waste: Chemical waste should be properly disposed of as outlined by Environmental Health & Safety office. No chemicals should be poured down the drains
Waste containers should be properly marked as and have hazardous waste tag tied to them at all times.
In case of emergency: The rules set forth by Environmental Health and Safety must be always followed. If an emergency requires evacuation, you must leave the cleanroom immediately and do not stop to un-gown. Also you must inform other users before leaving.
Equipment: All users must be trained before using any equipment. All equipment use should be scheduled. If you are unable to use the scheduled time, you must update your reservation. Also, it is every users’ responsibility to properly operate and clean each piece of equipment that they use and report damaged or malfunctioning equipment.
Housekeeping: Every user is responsible for keeping the cleanrooms clean. They must clean your workspace before leaving. They should not leave or store items on or in equipment. They should never set liquids on any equipment, properly store all materials before leaving the cleanroom.
Control of microbial growth
Sterilization: Killing or removing all forms of microbial life (including endospores) in a material or an object. Heating is the most commonly used method of sterilization.
Disinfection: Reducing the number of pathogenic microorganisms to the point where they no longer cause diseases. Usually involves the removal of vegetative or non-endospore forming pathogens.
Disinfection Methods & Terms
Disinfection may use physical or chemical methods. Several common terminology that is used with reference to disinfection are:
- Disinfectant: Applied to inanimate objects.
- Antiseptic: Applied to living tissue (antisepsis).
- Degerming: Mechanical removal of most microbes in a limited area. Example: Alcohol swab on skin.
- Sanitization: Use of chemical agent on food-handling equipment to meet public health standards and minimize chances of disease transmission. E.g: Hot soap & water.
- Sepsis: Comes from Greek for decay or putrid. Indicates bacterial contamination.
- Asepsis: Absence of significant contamination.
- Aseptic techniques are used to prevent contamination of surgical instruments, medical personnel, and the patient during surgery. Aseptic techniques are also used to prevent bacterial contamination in food industry.
Several factors can influence the effectiveness of antimicrobial treatment.
- Number of Microbes: The more microbes present, the more time it takes to eliminate population.
- Types of Microbes: Endospores are very difficult to destroy. Vegetative pathogens vary widely in susceptibility to different methods of microbial control.
- Environmental influences: Presence of organic material (blood, feces, saliva) tends to inhibit antimicrobials, pH etc.
- Time of Exposure: Chemical antimicrobials and radiation treatments are more effective at longer times.
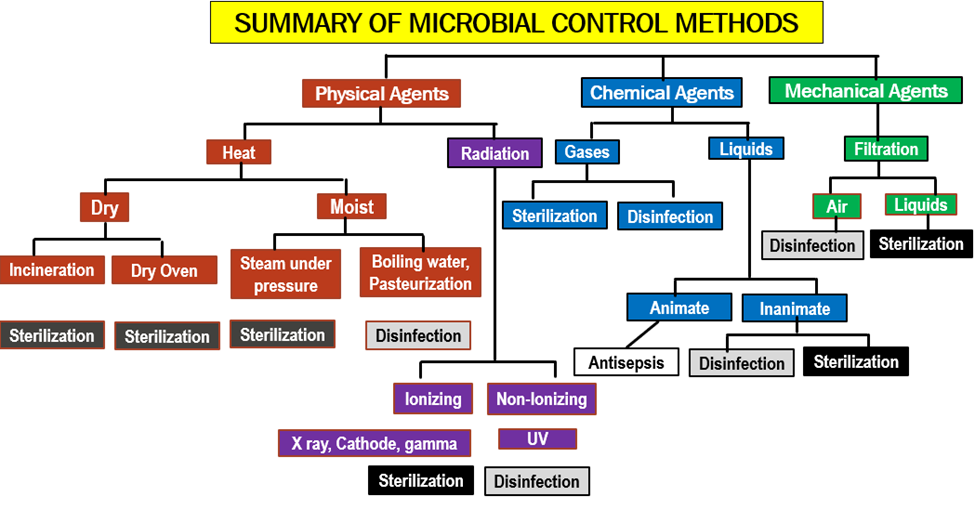
Laboratory Waste: There are different type of waste that labs can generate such as
- Normal Municipal waste (general)
- Recyclable waste
- Broken Glass
- Biological / Medical waste
- Chemical waste
- Sharps, Broken Glass
- Radioactive material waste
- Electronic and computer waste
Hazardous waste is any waste that directly or indirectly represents a threat to human health or to the environment by one or more of the following ways.
- Explosion or fire
- Infections, pathogens, parasites or their vectors
- Chemical instability, reactions or corrosion
- Acute or chronic toxicity
- Cancer, mutations or birth defects
- Toxicity or damage to the ecosystems or natural resources
- Accumulation in the biological food chain, persistence in the environment or multiple effects
Types of Biohazardous waste
- Sharps – Have the ability to cut or puncture.
- Pasteur pipettes
- Syringes with needles
- Needles
- Razor Blades
- Microscope slides
- Liquids – Pourable Wastes
- Stocks
- Media
- Blood
- Aspirated Liquid Wastes
- Dry Solid- No pourable liquids!
- Contaminated Containers such as:
- Petri Dishes
- Conical Tubes
- Contaminated Transfer Devices
- Pipette Tips
- Plastic Pipettes
- Contaminated Containers such as:
Waste Collection
- Dry Solids (No sharps!)
- Primary Containment: Collect dry, solid waste in a “red bag”. The red bag must have the international biohazard symbol, the word “biohazard” and a label.
- Dry Solids (No sharps!)
- Secondary Containment:
The red bag (primary containment) must be stored in a rigid container with a lid that is resistant to leaks and punctures. The red bag must be kept in the secondary container during use, storage, and transport.
- Sharps
- Collect in a rigid puncture and leak resistant properly labeled container.
- The words “Biohazardous waste”
- Must have International Biological Hazard symbol
- Collect in a rigid puncture and leak resistant properly labeled container.
- Biohazardous Liquid Waste (Temporary Storage)
- Collect in a non-breakable container with lid and labeled with the international biohazard symbol and the word “Biohazard”.
- The container needs to be in secondary containment.
- Biohazardous Liquid Waste Disposal
- Treat the liquid by disinfecting it with a 10% bleach solution.
- Let the solution stand for 20 minutes.
- Discard down sink drain, then flush with water.