Cancer and the Cell Cycle
Overview
By the end of this section, you will be able to:
- Describe how cancer is caused by uncontrolled cell growth
- Understand how proto-oncogenes are normal cell genes that, when mutated, become oncogenes
- Describe how tumor suppressors function
- Explain how mutant tumor suppressors cause cancer
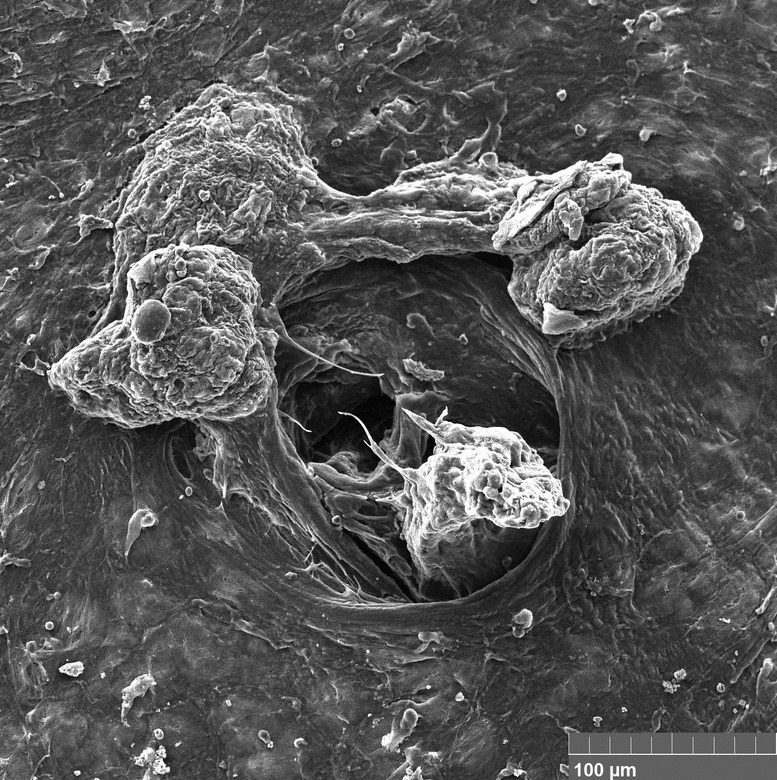
Opening Photo: Scanning electron micrograph. Pore of the lymphatic system in cancer. Tumor cells in the lumen of the pore and on the surface of the peritoneum. Credit: GANTSEV SHAMIL via Wikimedia Commons [CC BY-SA 4.0 (https://creativecommons.org/licenses/by-sa/4.0)]
By the end of this section, you will be able to:
- Define contact inhibition.
- Explain how cancer is caused by uncontrolled cell division
- Understand how proto-oncogenes are normal cell genes that, when mutated, become oncogenes
- Describe how tumor suppressors function to stop the cell cycle until certain events are completed
- Explain how mutant tumor suppressors cause cancer
- Define tumor, benign, malignant, metatasis, and apoptosis.
Does cell cycle control matter? If you ask an oncologist – a doctor who treats cancer patients – she or he will likely answer with a resounding yes.
Cancer is basically a disease of uncontrolled cell division. Its development and progression are usually linked to a series of changes in the activity of cell cycle regulators. For example, inhibitors of the cell cycle keep cells from dividing when conditions aren’t right, so too little activity of these inhibitors can promote cancer. Similarly, positive regulators of cell division can lead to cancer if they are too active. In most cases, these changes in activity are due to mutations in the genes that encode cell cycle regulator proteins.
Here, we’ll look in more detail at what's wrong with cancer cells. We'll also see how abnormal forms of cell cycle regulators can contribute to cancer.
What's Wrong with Cancer Cells?
Cancer cells behave differently than normal cells in the body. Many of these differences are related to cell division behavior.
For example, cancer cells can multiply in culture (outside of the body in a dish) without any growth factors, or growth-stimulating protein signals, being added. This is different from normal cells, which need growth factors to grow in culture.
Cancer cells may make their own growth factors, have growth factor pathways that are stuck in the "on" position, or, in the context of the body, even trick neighboring cells into producing growth factors to sustain 1.
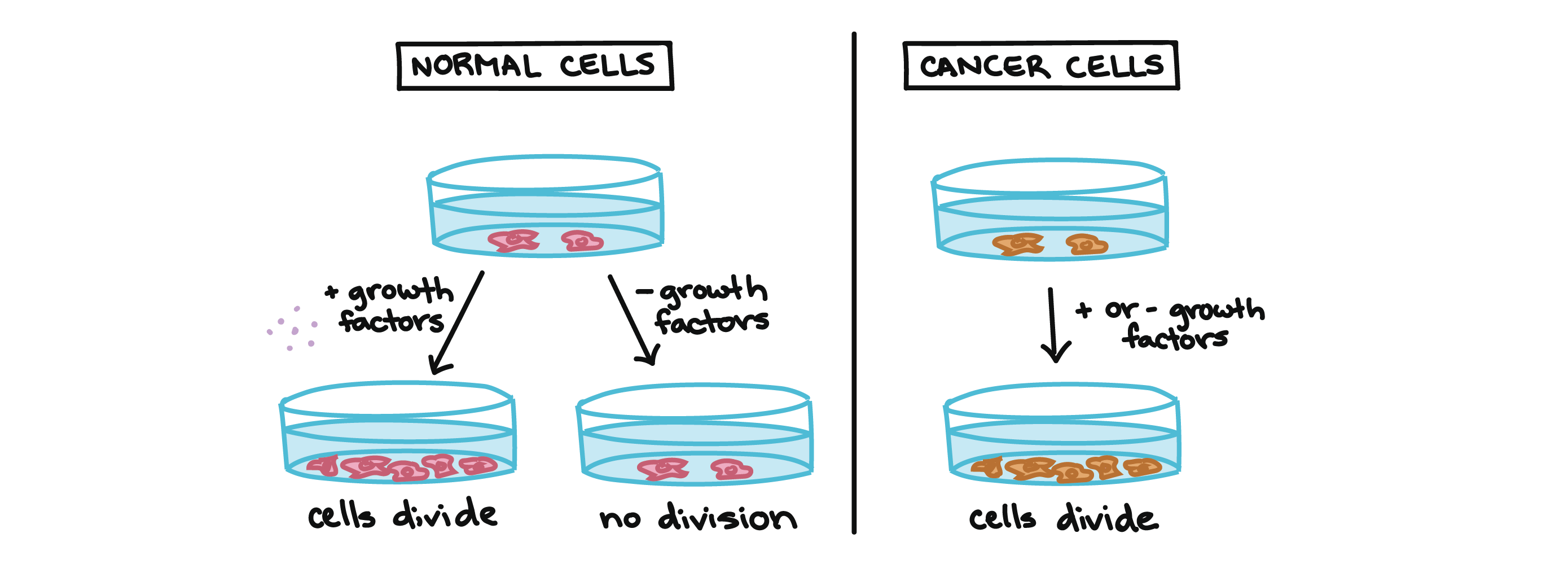
Cancer cells also ignore signals that should cause them to stop dividing. For instance, when normal cells grown in a dish are crowded by neighbors on all sides, they will no longer divide. Cancer cells, in contrast, keep dividing and pile on top of each other in lumpy layers.
The environment in a dish is different from the environment in the human body, but scientists think that the loss of contact inhibition in plate-grown cancer cells reflects the loss of a mechanism that normally maintains tissue balance in the body2.
Another hallmark of cancer cells is their "replicative immortality," a fancy term for the fact that they can divide many more times than a normal cell of the body. In general, human cells can go through only about 40-60 rounds of division before they lose the capacity to divide, "grow old," and eventually die3.
Cancer cells can divide many more times than this, largely because they express an enzyme called telomerase, which reverses the wearing down of chromosome ends that normally happens during each cell division4.
Cancer cells are also different from normal cells in other ways that aren’t directly cell cycle-related. These differences help them grow, divide, and form tumors. For instance, cancer cells gain the ability to migrate to other parts of the body, a process called metastasis, and to promote growth of new blood vessels, a process called angiogenesis (which gives tumor cells a source of oxygen and nutrients). Cancer cells also fail to undergo programmed cell death, or apoptosis, under conditions when normal cells would (e.g., due to DNA damage). In addition, emerging research shows that cancer cells may undergo metabolic changes that support increased cell growth and division5.
How cancer develops
Cells have many different mechanisms to restrict cell division, repair DNA damage, and prevent the development of cancer. Because of this, it’s thought that cancer develops in a multi-step process, in which multiple mechanisms must fail before a critical mass is reached and cells become cancerous. Specifically, most cancers arise as cells acquire a series of mutations (changes in DNA) that make them divide more quickly, escape internal and external controls on division, and avoid programmed cell death6.
How might this process work? In a hypothetical example, a cell might first lose activity of a cell cycle inhibitor, an event that would make the cell’s descendants divide a little more rapidly. It’s unlikely that they would be cancerous, but they might form a benign tumor, a mass of cells that divide too much but don’t have the potential to invade other tissues (metastasize)7.
Over time, a mutation might take place in one of the descendant cells, causing increased activity of a positive cell cycle regulator. The mutation might not cause cancer by itself either, but the offspring of this cell would divide even faster, creating a larger pool of cells in which a third mutation could take place. Eventually, one cell might gain enough mutations to take on the characteristics of a cancer cell and give rise to a malignant tumor, a group of cells that divide excessively and can invade other tissues7.
Proto-oncogenes
Positive cell cycle regulators may be overactive in cancer. The genes that code for the positive cell-cycle regulators are called proto-oncogenes. Proto-oncogenes are normal genes that, when mutated, become oncogenes—genes that cause a cell to become cancerous. Consider what might happen to the cell cycle in a cell with a recently acquired oncogene. In most instances, the alteration of the DNA sequence will result in a less functional (or non-functional) protein. The result is detrimental to the cell and will likely prevent the cell from completing the cell cycle; however, the organism is not harmed because the mutation will not be carried forward. If a cell cannot reproduce, the mutation is not propagated and the damage is minimal. Occasionally, however, a gene mutation causes a change that increases the activity of a positive regulator. For example, a mutation that allows Cdk, a protein involved in cell-cycle regulation, to be activated before it should be could push the cell cycle past a checkpoint before all of the required conditions are met. If the resulting daughter cells are too damaged to undertake further cell divisions, the mutation would not be propagated and no harm comes to the organism. However, if the atypical daughter cells are able to divide further, the subsequent generation of cells will likely accumulate even more mutations, some possibly in additional genes that regulate the cell cycle.
The Cdk example is only one of many genes that are considered proto-oncogenes. In addition to the cell-cycle regulatory proteins, any protein that influences the cycle can be altered in such a way as to override cell-cycle checkpoints. Once a proto-oncogene has been altered such that there is an increase in the rate of the cell cycle, it is then called an oncogene.
Tumor Suppressor Genes
Like proto-oncogenes, many of the negative cell-cycle regulatory proteins were discovered in cells that had become cancerous. Tumor suppressor genes are genes that code for the negative regulator proteins, the type of regulator that—when activated—can prevent the cell from undergoing uncontrolled division. The collective function of the best-understood tumor suppressor gene proteins, retinoblastoma protein (RB1), p53, and p21, is to put up a roadblock to cell-cycle progress until certain events are completed. A cell that carries a mutated form of a negative regulator might not be able to halt the cell cycle if there is a problem.
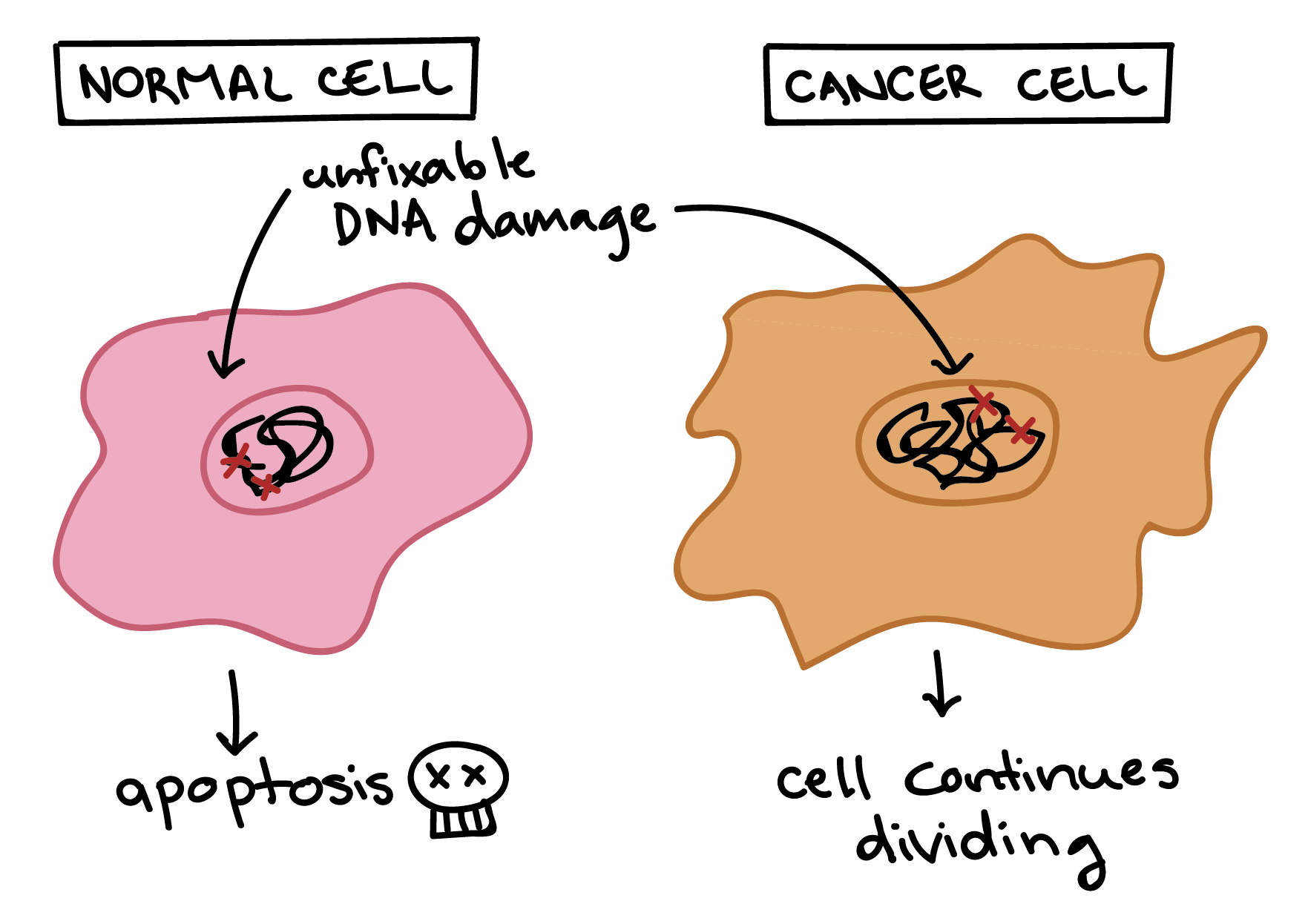
Mutated p53 genes have been identified in more than half of all human tumor cells. This discovery is not surprising in light of the multiple roles that the p53 protein plays at the G1 checkpoint. The p53 protein activates other genes whose products halt the cell cycle (allowing time for DNA repair), activates genes whose products participate in DNA repair, or activates genes that initiate cell death when DNA damage cannot be repaired. A damaged p53 gene can result in the cell behaving as if there are no mutations (Figure 6.8). This allows cells to divide, propagating the mutation in daughter cells and allowing the accumulation of new mutations. In addition, the damaged version of p53 found in cancer cells cannot trigger cell death.
Art Connection

Human papillomavirus can cause cervical cancer. The virus encodes E6, a protein that binds p53. Based on this fact and what you know about p53, what effect do you think E6 binding has on p53 activity?
- E6 activates p53
- E6 inactivates p53
- E6 mutates p53
- E6 binding marks p53 for degradation
The loss of p53 function has other repercussions for the cell cycle. Mutated p53 might lose its ability to trigger p21 production. Without adequate levels of p21, there is no effective block on Cdk activation. Essentially, without a fully functional p53, the G1 checkpoint is severely compromised and the cell proceeds directly from G1 to S regardless of internal and external conditions. At the completion of this shortened cell cycle, two daughter cells are produced that have inherited the mutated p53 gene. Given the non-optimal conditions under which the parent cell reproduced, it is likely that the daughter cells will have acquired other mutations in addition to the faulty tumor suppressor gene. Cells such as these daughter cells quickly accumulate both oncogenes and non-functional tumor suppressor genes. Again, the result is tumor growth.
Link to Learning

Watch an animation of how cancer results from errors in the cell cycle.
Section Summary
Cancer is the result of unchecked cell division caused by a breakdown of the mechanisms that regulate the cell cycle. The loss of control begins with a change in the DNA sequence of a gene that codes for one of the regulatory molecules. Faulty instructions lead to a protein that does not function as it should. Any disruption of the monitoring system can allow other mistakes to be passed on to the daughter cells. Each successive cell division will give rise to daughter cells with even more accumulated damage. Eventually, all checkpoints become nonfunctional, and rapidly reproducing cells crowd out normal cells, resulting in a tumor or leukemia (blood cancer).
Art Connections
Figure Human papillomavirus can cause cervical cancer. The virus encodes E6, a protein that binds p53. Based on this fact and what you know about p53, what effect do you think E6 binding has on p53 activity?
A. E6 activates p53
B. E6 inactivates p53
C. E6 mutates p53
D. E6 binding marks p53 for degradation
Hint:
Figure D. E6 binding marks p53 for degradation.
Review Questions
___________ are changes to the order of nucleotides in a segment of DNA that codes for a protein.
A. Proto-oncogenes
B. Tumor suppressor genes
C. Gene mutations
D. Negative regulators
Hint:
C
A gene that codes for a positive cell cycle regulator is called a(n) _____.
A. kinase inhibitor.
B. tumor suppressor gene.
C. proto-oncogene.
D. oncogene.
Hint:
C
A mutated gene that causes a cell to be cancerous is called a(n) _____.
A. kinase inhibitor.
B. tumor suppressor gene.
C. proto-oncogene.
D. oncogene.
Hint:
D
_______ can prevent the cell from undergoing uncontrolled division.
A. Kinase inhibitors
B. Oncogenes
C. Proto-oncogenes
D. Tumor suppressor genes
Hint:
D
Free Response
Outline the steps that lead to a cell becoming cancerous.
Hint:
If one of the genes that produces regulator proteins becomes mutated, it produces a malformed, possibly non-functional, cell cycle regulator, increasing the chance that more mutations will be left unrepaired in the cell. Each subsequent generation of cells sustains more damage. The cell cycle can speed up as a result of the loss of functional checkpoint proteins. The cells can lose the ability to self-destruct and eventually become “immortalized.”
Explain the difference between a proto-oncogene and a tumor suppressor gene.
Hint:
A proto-oncogene is a segment of DNA that codes for one of the positive cell cycle regulators. If that gene becomes mutated so that it produces a hyperactivated protein product, it is considered an oncogene. A tumor suppressor gene is a segment of DNA that codes for one of the negative cell cycle regulators. If that gene becomes mutated so that the protein product becomes less active, the cell cycle will run unchecked. A single oncogene can initiate abnormal cell divisions; however, tumor suppressors lose their effectiveness only when both copies of the gene are damaged.
p53 can trigger apoptosis if certain cell cycle events fail. How does this regulatory outcome benefit a multicellular organism?
Hint:
If a cell has damaged DNA, the likelihood of producing faulty proteins is higher. The daughter cells of such a damaged parent cell would also produce faulty proteins that might eventually become cancerous. If p53 recognizes this damage and triggers the cell to self-destruct, the damaged DNA is degraded and recycled. No further harm comes to the organism. Another healthy cell is triggered to divide instead.
Attributions
This module is a derivative of OpenStax Biology's (2e), and Khan Academy's article Cancer and the Cell Cycle.
Openstax Biology license: (CC BY 3.0). Download the original article for free at http://cnx.org/contents/185cbf87-c72e-48f5-b51e-f14f21b5eabd@9.85.
Khan Acadmy License: CC BY-NC-SA 4.0(Opens in a new window)(Opens in a new window)(Opens in a new window) license. Download the original article for free at https://www.khanacademy.org/science/biology/cellular-molecular-biology/stem-cells-and-cancer/a/cancer.
Works cited:
Hanahan, D. and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 647. http://dx.doi.org/10.1016/j.cell.2011.02.013.647
Hanahan, D. and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 649. http://dx.doi.org/10.1016/j.cell.2011.02.013.
Bartlett, Zane. (2014, November 14). The Hayflick limit. In The Embryo Project Encyclopedia. Retrieved from https://embryo.asu.edu/pages/hayflick-limit.
Hanahan, D. and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 651. http://dx.doi.org/10.1016/j.cell.2011.02.013.647
Hanahan, D. and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell, 144(5), 647, 659-660. http://dx.doi.org/10.1016/j.cell.2011.02.013.647
Vogelstein, B. and Kinzler, K. W. (2004). Cancer genes and the pathways they control. Nature Medicine, 10, 790. http://dx.doi.org/10.1038/nm1087.
Reece, J. B., Urry, L. A., Cain, M. L., Wasserman, S. A., Minorsky, P. V., and Jackson, R. B. (2011). The cell cycle. In Campbell biology (10th ed.). San Francisco, CA: Pearson, 247
Vogelstein, B. and Kinzler, K. W. (2004). Cancer genes and the pathways they control. Nature Medicine, 10, 789. http://dx.doi.org/10.1038/nm1087.